




- Arbeitsgruppen
- Klinisches Studienzentrum
- Sektion Kardio-Onkologie
- Section of Molecular and Translational Cardiology
- Klaus-Tschira-Institut für Computerkardiologie
- Heisenberg Professur für Immunkardiologie (W3)
- Jun. Professur für Künstliche Intelligenz in der Kardiovaskulären Medizin
- Pregnancy Heart Team: Kardiologie der Schwangerschaft
- DZHK-Standort Heidelberg/Mannheim
- Heidelberg CardioBiobank (HCB)
- Scientific Management and Coordination Unit (SMCU)
AG Extracellular Matrix and Integrins in Cardiovascular Disease
Integrin signaling and heart failure
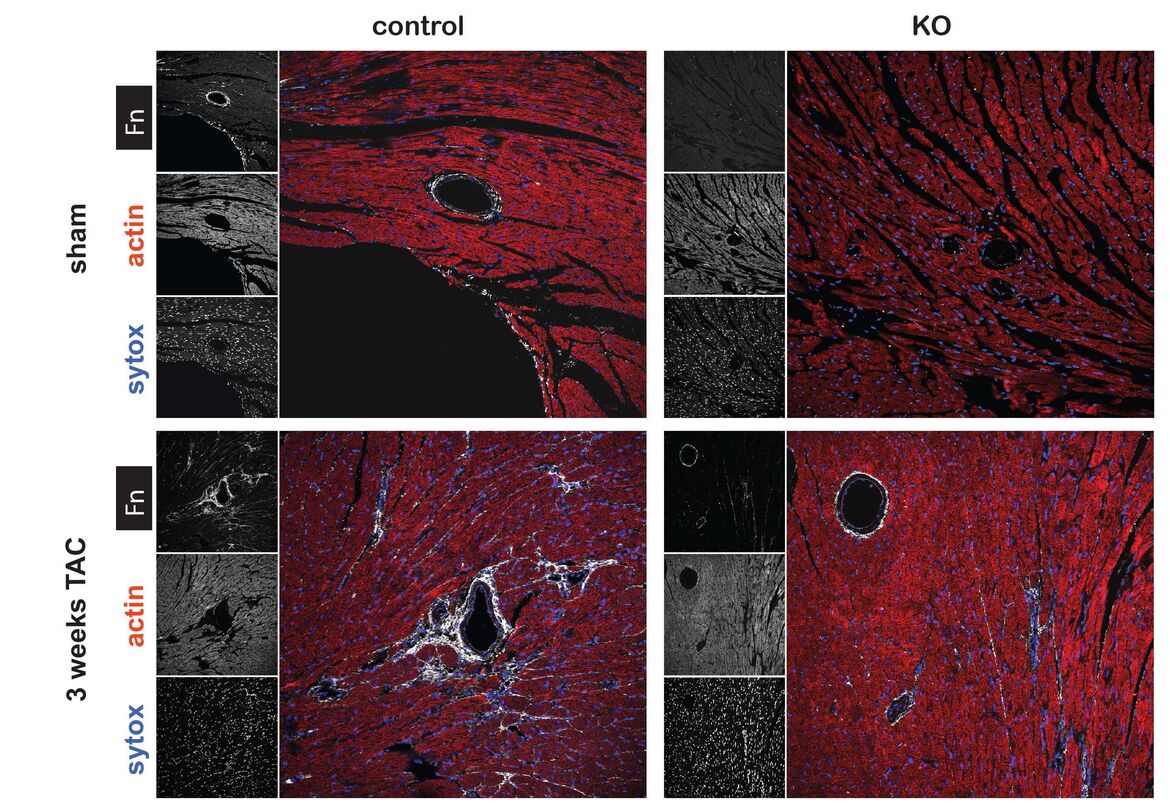
Integrins are heterodimeric surface receptors consisting of one α-chain and one ß-chain. There are 18 α-chains and 8 ß-chains, resulting in 24 combinations that define ligand specificity. Binding of the respective ligand activates signaling cascades, relaying information from the environment to the inside of the cell (outside-in signaling) and regulating pivotal biological functions such as proliferation, growth, differentiation, or apoptosis. By establishing a Tamoxifen (Tx) inducible knockout mouse, we have demonstrated that the extracellular matrix glycoprotein fibronectin (Fn), activates critical survival signals via ß1 integrin following myocardial infarction (1). In a second study, we have shown that fibronectin is essential for pathological cardiac hypertrophy upon pressure-overload and ultimately leads to heart failure. Whereas, physiological growth due to endurance exercise training is independent of fibronectin (2). In a DFG funded project we are currently dissecting the role of integrin receptors in cardiac hypertrophy in vitro and in vivo by applying the conditional Tx-driven Cre-lox technology.
Integrin activation and atherosclerosis
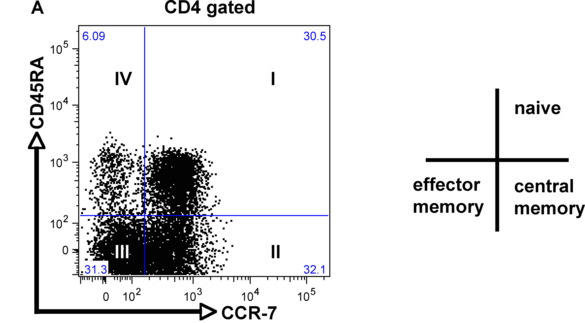
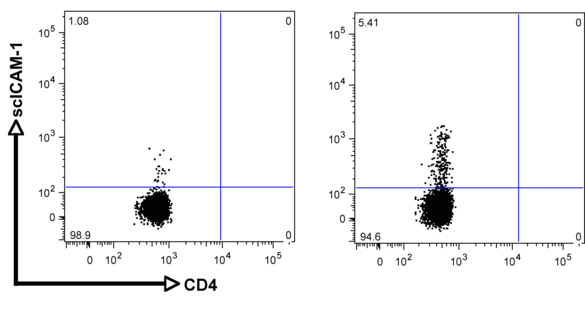
Although integrins are highly expressed on the cell surface, they do not bind their respective ligand unless activated from within the cell (inside-out signaling). This tightly controlled process is pivotal during leukocyte migration and recruitment to the site of inflammation, as well as metastasis of cancer cells. ß2 integrin activation on T cells is also a critical step during the process of extravasation and recruitment of T cells within the atherosclerotic plaque. We recently established a novel flow-cytometry based whole blood application to quantify functional ß2 integrin activation (induction of affinity and avidity) on T cell subpopulations. With this technique, we can demonstrate that ß2 integrin activation on T cells in patients with acute coronary syndrome (ACS), precedes future cardiovascular events and provides additional prognostic information over established biomarkers such as cardiac troponin (cTnT) or high-sensitive C-reactive protein (hsCRP) (6). The aim of this project is to characterize differentially regulated signaling cascades in T cell subpopulations and verify the relevance of these pathways for the progression of atherosclerosis in established mouse models.
Metabolic reprogramming during cardiac hypertrophy and heart failure
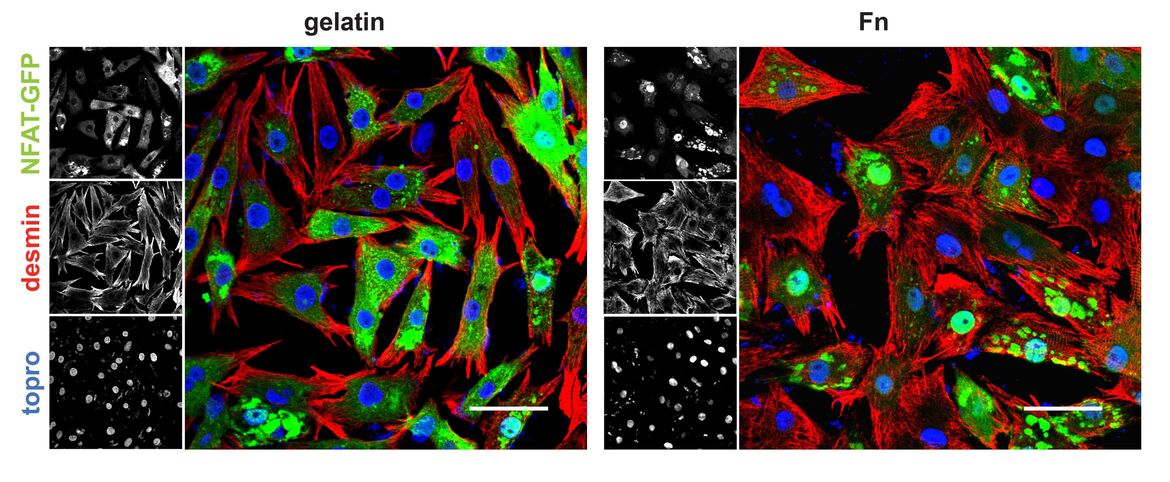
Cardiac hypertrophy occurs in response to increased workload and can diverge into two modalities, pathological or physiological growth. Pathological remodeling, upon aortic stenosis or hypertension, is associated with increased mortality. Physiological growth, through endurance training, has no detrimental impact upon survival and is even beneficial for the pathologically stressed heart. A notable characteristic defining hypertrophy is alterations in metabolism. Ordinarily, the heart utilizes fatty acids as a main fuel source, however, pathological remodeling and deterioration of cardiac function correlates with ATP deprivation and a fuel shift away from fatty acid oxidation (FAO) towards glucose metabolism. In contrast, physiological hypertrophy is accompanied by increases in FAO rates, the capacity for mitochondrial respiration, and ATP synthesis. In this project we analyze effects of extracellular cues for hypertrophic remodeling and examine transcription factor circuits regulating metabolic reprogramming. Taking advantage of siRNA and the cre-lox technology for cardiomyocyte specific knockouts, experiments are performed in vivo and in vitro to assess causality of the respective proteins in the heart.
Scientific methodology
A broad spectrum of state-of-the-art techniques are applied in the laboratory including employing multi-color flow cytometry for phenotyping and functional analysis of integrin activation, cellular apoptosis, cell cycle and proliferation. Other techniques utilized in the lab are high throughput screening for cell size analysis, Seahorse technology for mitochondrial respiration, confocal microscopy, real time PCR, immunoblot, immunhiostochemistry, ELISAs, etc. Primary cells (cardiomyocytes, hematopoietic stem cells, t cells) and cell lines (macrophages, HEk293s, HeLa) are used for in vitro experiments. Applications in vitro include siRNA treatment, adenoviral, or lentiviral infection. Relevance of our in vitro findings are confirmed in in vivo mouse models (myocardial infarction, transverse aortic banding, endurance exercise, atherosclerosis model), including genetic knockout mice. To evaluate cardiac function small animal echocardiography is applied.
Collaborations
Heidelberg
- Prof. Dr. J. Backs, Molecular Cardiology and Epigenetics
- Prof. Dr. M. Boutros, DKFZ
- Prof. Dr. C. Dieterich, Cardiology
- Prof. Dr. F. Leuschner, Cardiology
- Prof. Dr. B. Meder, Cardiology
- Dr. M. Völkers, Cardiology
- Prof. Dr. Y. Samstag, PD Dr. G. Wabnitz, Institute of Immunology
- EMBL Genomics Core Facility
National
- Prof. Dr. S. Herzig, Helmholtz Centre Munich, Germany
- Prof. Dr. R. Fässler, Max Planck Institute, Martinsried, Germany
- Dr. Braren, University Hospital Eppendorf, Hamburg, Germany
International
- Prof. Dr. A. Sonnenberg, Amsterdam, Netherland Cancer Institute
- Prof. Dr. M. Sussman, San Diego, Biology Department, San Diego State University, California, USA
Funding
DFG, DZHK, ASPHIRE Award (Pfizer), Department of Cardiology HD
Representative Publications (selection)
- Konstandin, M.H., Toko, H., Gastelum, G.M., Quijada, P.J., De La Torre, A., Quintana, M., Collins, B., Din, S., Avitabile, D., Volkers, M.J., et al.. Fibronectin is Essential for Reparative Cardiac Progenitor Cell Response Following Myocardial Infarction. Circulation research. 2013. Jul 5;113(2):115-25.
- Volkers M, Konstandin MH, Doroudgar S, Toko H, Quijada P, Din S, Joyo A, Ornelas L, Samse K, Thuerauf DJ, Gude N, Glembotski CC, Sussman MA. Mtorc2 protects the heart from ischemic damage. Circulation. 2013. Nov 5;128(19):2132-44.
- Konstandin MH, Volkers M, Collins B, Quijada P, Quintana M, De La Torre A, Ormachea L, Din S, Gude N, Toko H, Sussman MA. Fibronectin contributes to pathological cardiac hypertrophy but not physiological growth. Basic research in cardiology. 2013;108:375
- Toko, H., Konstandin, M.H., Doroudgar, S., Ormachea, L., Joyo, E., Joyo, A.Y., Din, S., Gude, N.A., Collins, B., Volkers, M., et al. Regulation of cardiac hypertrophic signaling by prolyl isomerase pin1. Circulation Research. 2013. 112, 1244-1252.
- Din S, Mason M, Volkers M, Johnson B, Cottage CT, Wang Z, Joyo AY, Quijada P, Erhardt P, Magnuson NS, Konstandin MH, Sussman MA. Pim-1 preserves mitochondrial morphology by inhibiting dynamin-related protein 1 translocation. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:5969-5974
- Konstandin MH, Aksoy H, Wabnitz GH, Volz C, Erbel C, Kirchgessner H, Giannitsis E, Katus HA, Samstag Y, Dengler TJ. Beta2-integrin activation on T cell subsets is an independent prognostic factor in unstable angina pectoris. Basic Res Cardiol. 2009 May;104(3):341-51.
- Din S, Konstandin MH, Johnson B, Emathinger JM, Volkers M, Toko H, Collins B, Ormachea L, Samse KM, Kubli DA, De La Torre A, Kraft AS, Gustafsson AB, Kelly DP, Sussman MA. Metabolic Dysfunction Consistent with Premature Aging Results from Deletion of Pim Kinases. Circulation Research. 2014 Jul 18;115(3):376-87.
- Arbeitsgruppen
- Klinisches Studienzentrum
- Sektion Kardio-Onkologie
- Section of Molecular and Translational Cardiology
- Klaus-Tschira-Institut für Computerkardiologie
- Heisenberg Professur für Immunkardiologie (W3)
- Jun. Professur für Künstliche Intelligenz in der Kardiovaskulären Medizin
- Pregnancy Heart Team: Kardiologie der Schwangerschaft
- DZHK-Standort Heidelberg/Mannheim
- Heidelberg CardioBiobank (HCB)
- Scientific Management and Coordination Unit (SMCU)
Kontakt
Arbeitsgruppenleiter
