Arbeitsgruppe Translationale Medizin
Unser Labor wurde im Rahmen einer Kooperation zwischen der Max-Planck Gesellschaft und der Universität Heidelberg eingerichtet. Ziel dieser Kooperation ist es, klinisch-relevante Fragen mit Hilfe von neuesten Grundlagentechniken zu beantworten. Unser Partner an der Max-Planck Gesellschaft ist Reinhard Fässler und seine Abteilung (Molekulare Medizin), mit denen eine enge Zusammenarbeit besteht.
Der Fokus unseres Labors ist die Untersuchung der Rolle der extrazellulären Matrix und deren Interaktion mit den umgebenden Zellen in der normalen Physiologie und bei verschiedenen Erkrankungen. Dies kann in zwei große Kategorien unterteilt werden:
- die Rolle der Matrix selbst und
- die Funktion der Matrixrezeptoren der Zellen.
Fibronektin ist ein essentieller Bestandteil der extrazellulären Matrix, mit dem wir uns beschäftigen. Knockout-Mäuse ohne Fibronektin sterben bereits in den Frühstadien der embryonalen Entwicklung. Auch postnatal hat dieses Protein vielfältige Aufgaben. Unser Forschungsprogramm beinhaltet die Untersuchung der Funktion des Fibronektins in verschiedenen Geweben (z.B. Knochen), der Rolle des Fibronektins bei verschiedenen Erkrankungen (Leberfibrose, Nierenerkrankungen und Tumorerkrankungen), sowie der Rezeptoren, die diese Auswirkungen vermitteln.
Die Zellen interagieren mit den Matrixproteinen durch verschiedene Rezeptoren, darunter die Integrine. Eine solche Bindung führt zu einer Reihe intrazellulärer Ereignisse, die durch Signalproteine vermittelt werden. In unserer Arbeit untersuchen wir die Auswirkung der Ausschaltung solcher Moleküle sowohl pränatal als auch postnatal.Wir verwenden verschiedene Techniken, wie z.B. Protein-Biochemie (Western, ELISA, Massenspektrometrie), zelluläre und molekulare Biologie (PCR und quantitative real-time PCR), verschiedene Färbungsmethoden und Analysen (Cryoschnitte, Lasermikrodissektion, Immunhistochemie und in situ Hybridisierung) und verschiedene Techniken zur Untersuchung der Knochen (pQCT, microCT, statische und dynamische Histomorphometrie). Ferner führen wir Untersuchungen in genetisch veränderten Mäusen (auch konditionellen Knockout-Mäusen) und in vivo Experimente durch.
Durch die enge Zusammenarbeit einer universitären Einrichtung mit einem Grundlagenforschungsinstitut bietet unser Labor die Möglichkeit, patientenorientierte Fragestellungen mittels der neuesten Forschungstechniken zu bearbeiten.
Aktuelle Liste der Publikationen
Weitere Informationen (English)
Lebererkrankungen
Viele Lebererkrankungen führen zur Bildung einer Fibrose, die letztendlich eine Zirrhose und Leberversagen verursacht. Die Fibrose ist Folge der Ansammlung von Molekülen der extrazellulären Matrix inkl. Fibronektin und Kollegen Typ I. Diese Moleküle werden u.a. von den Sternzellen der Leber produziert, wenn sie mit dem Zytokin TGF-b stimuliert werden.
Unsere Gruppe zeigte erstmals, dass die Isoformen des Fibronektins erhöht sind bei Patienten mit chronisch cholestatischen Lebererkrankungen. Die Beteiligung einer Isoform des Fibronektins (nämlich der onkofötalen Isoform) bei der assoziierten sekundären Osteoporose konnte ebenfalls festgestellt werden (Kawelke et al. https://www.biochem.mpg.de/en/rg/nakchbandi/publications) (siehe unten unter Osteoporose). Des Weiteren konnten wir bestätigen, dass bei einem Drittel der Patienten mit chronischer Hepatitis C Infektion das Ausmaß der entstehenden Fibrose vorhergesagt werden kann, da bei diesen Patienten die Fibronektin-Isoformen beeinträchtigt sind (Hackl et al. https://www.biochem.mpg.de/en/rg/nakchbandi/publications).
Basierend auf diesen Arbeiten haben wir damit begonnen, die Auswirkungen einer Ausschaltung des Fibronektins in den Sternzellen zu untersuchen. Da ein vollständiger Fibronektin-Knockout bereits in utero letal ist, musste dies mittels des Cre-loxP Systems erfolgen. In diesem System wird das Fibronektin-Gen mit zwei sogenannten loxP Stellen jeweils oberhalb und unterhalb des ATG-Startkodons versehen. Zellen, deren Fibronektin-Gen ausgeschaltet werden soll, wird die Cre-Rekombinase unter der Kontrolle eines Promoters inseriert, welcher spezifisch in dem zu untersuchenden Zelltypen aktiviert wird. Die Cre-Rekombinase ist dann in der Lage, das Gen an den loxP Stellen zu schneiden und die Schnittstellen wieder miteinander zu verbinden. Dies führt zu einem defekten Gen und letztendlich zum Verlust des Fibronektins.
Mäuse, die nicht dazu in der Lage waren, in den Sternzellen Fibronektin zu produzieren, zeigten schon unstimuliert eine deutliche Verstärkung der Fibrosebildung, was zunächst sehr überraschend war (Abbildung). Daraus lässt sich schließen, dass die Sternzellen in Abwesenheit von Fibronektin überstimuliert sind. Wir gehen davon aus, dass die TGF-b Produktion, bzw. Speicherung Fibronektin benötigt (Kawelke et al. Link: https://www.ncbi.nlm.nih.gov/pubmed/22140539).
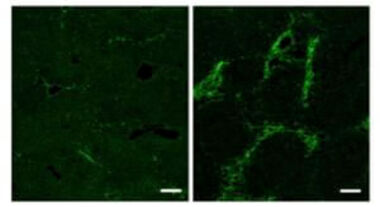
Weitere Arbeiten auf diesem Gebiet beinhalten die Untersuchung der Fibrosebildung bei Mäusen, bei denen Fibronektin in mutierter Form produziert wird und daher nicht mit RGD-bindenden Integrinen interagieren kann. Auch untersuchen wir, wie man die Matrixansammlung, und somit die Fibrose vermindern kann. Ergebnisse dazu wurden bereits publiziert (Altrock et al. Link: http://www.sciencedirect.com/science/article/pii/S016882781400405X?via%3Dihub).
Nierenerkrankungen
Diabetes mellitus ist durch einen chronisch erhöhten Blutzucker (Hyperglykämie) gekennzeichnet. Eine wichtige Komplikation ist die Diabetes-bedingte Nierenerkrankung, die in den USA und Westeuropa die häufigste Ursache für eine Niereninsuffizienz. Die diabetische Nephropathie ist im Mausmodell durch eine Ausbreitung der Mesangialmatrix und eine Verdickung der glomerulären Basalmembran gekennzeichnet. Der Ausbreitung und Verdickung liegen die Einlagerung des Fibronektins und anderer Matrixproteine zugrunde.
Vorarbeiten haben gezeigt, dass injiziertes Fibronektin durch die Blutzirkulation in die Niere gelangt und in der Mesangialmatrix der Glomeruli eingelagert wird. Basierend auf diesen Arbeiten haben wir damit begonnen, die Auswirkungen einer Ausschaltung des Fibronektins in der Blutbahn und in den Mesangialzellen zu untersuchen. Da ein vollständiger Fibronektin-Knockout bereits in utero letal ist, musste dies mittels des Cre-loxP Systems erfolgen. In diesem System wird das Fibronektin-Gen mit zwei sogenannten loxP Stellen jeweils oberhalb und unterhalb des ATG-Startkodons versehen. Zellen, dessen Fibronektin-Gen ausgeschaltet werden soll, wird die Cre-Rekombinase unter der Kontrolle eines Promoters inseriert, welcher spezifisch in dem zu untersuchenden Zellty-pen aktiviert wird. Die Cre-Rekombinase ist dann in der Lage, das Gen an den loxP Stellen zu schneiden und die Schnittstellen wieder miteinander zu verbinden. Dies führt zu einem defekten Gen und letztendlich zum Verlust des Fibronektins.
In konditionellen Knockout-Mäusen, bei denen das Plasmafibronektin bzw. das Fibronektin der Mesangialzellen und das Plasmafibronektin zugleich ausgeschaltet wurden, induzierten wir Diabetes mellitus. Die Ausschaltung des Fibronektins hatte eine geringere Ausbreitung der Mesangialmatrix sowie eine geringere Mortalität der Tiere zur Folge. Interessanterweise schien das Plasmafibronektin alleine bereits grob ein Drittel der Ausdehnung des Mesangiums zu verursachen (Klemis et al. Link: http://www.sciencedirect.com/science/article/pii/S0085253816307062?via%3Dihub).
Weitere Arbeiten in diesem Gebiet beschäftigen sich mit der Interaktion der Mesangialzellen und der umgebenden Matrix, sowie dem Einfluss der Hyperglykämie auf die Matrixbildungseigenschaften des Fibronektins. Weiter wird die Interaktion des in der Niere gelagerten Fibronektins auf das Verhalten der Mesangialzellen untersucht. Ziel ist es, ein Verständnis des Einflusses von Diabetes bei der Bildung einer Matrix zu erzielen und daraus neue Therapieansätze zu entwickeln.
Tumorerkrankungen
Maligne Tumorerkrankungen sind in Deutschland nach Herz-Kreislauf-Erkrankungen mit ca. 25% aller Todesfälle die zweithäufigste Todesursache. In der Metastasierung liegt das eigentliche klinische Problem von Krebserkrankungen begründet, denn 90% aller krebsbezogenen Todesfälle sind auf die Bildung von Metastasen zurückzuführen.
Tumorzellen des Brust- und Prostatakrebses bilden häufig Absiedelungen im Knochenmark. Es wird vermutet, dass die Tumorzellen bereits vorhandene physiologische Stammzellnischen ihren Anforderungen entsprechend modifizieren. Im Knochenmark konnten zwei Nischen identifiziert werden, die das Wachstum hämatopoetischer Stammzellen begünstigen und womöglich auch von Tumorzellen auf unterschiedliche Weise genutzt werden. Es wird zwischen der vaskulären Nische, die im Bereich der sinusoidalen Blutgefäße des Knochenmarks angesiedelt ist, und der osteoblastischen Nische, die an der Grenzfläche zwischen Knochen und Knochenmark lokalisiert ist, differenziert. Somit könnte das Knochenmark im Falle des Brust- und Prostatakrebses als eine Art prämetastatische Nische fungieren. Neuere Erkenntnisse zeigen, dass tumorzellspezifische Faktoren entfernte Gewebe auf die Besiedelung durch Tumorzellen vorbereiten. Dabei wird Fibronektin im Bereich der prämetastatischen Nische vermehrt exprimiert.
Fibronektin der Mikroumgebung des Knochenmarks wurde im Mausmodell unter Verwendung des sequenzspezifischen Cre/loxP Rekombinationssystems ausgeschaltet. Unterschiedliche Zellen wurden somit ausgeschaltet und dies führte zu interessanten Effekt auf das Tumorwachstum, die uns erlauben ein besseres Verständnis der Krebsentwicklungs- und metastasierungsprozesse zu erlangen. Z.B. hatte die Fibronektin-Repression in der Zirkulation ein verlangsamtes Tumorwachstum zur Folge, welches auf eine verminderte Angiogenese zurückzuführen war. Im Gegensatz dazu, beeinträchtigte die osteoblastenspezifische Fibronektin-Deletion die frühen Entwicklungsstadien der Tumore. Aus diesen Arbeiten sind mehrere Publikationen entstanden. (von Au et al. Link: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3730044/; Rossnagl et al. Link: http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0094922; Rossnagl et al. Link: https://www.ncbi.nlm.nih.gov/pubmed/27653627).
Weitere Untersuchungen beschäftigen sich mit der Rolle des Fibronektins beim Einnisten der Tumorzellen in den metastatischen Nischen („Homing“) Rossnagl et al. Link: http://cancerres.aacrjournals.org/content/early/2017/10/24/0008-5472.CAN-16-3507.long). Die Anwendung von Proteomanalysen (mittels SILAC-Technologie) sollte Hinweise darauf geben, wie die frühen Stadien der Einnistung der Tumorzellen im Knochenmark beeinträchtigt werden können. Untersuchungen zur Immunantwort in Abwesenheit des Fibronektins haben bereits Aufschluss darüber geben, welche Möglichkeiten es gibt um das Krebswachstum auf neuartigen Wege zu bremsen bzw. zu stoppen.
Osteoporose
Unsere Gruppe konnte erstmals die Beteiligung einer Isoform des Fibronektins (nämlich der onkofötalen Isoform) bei der sekundären Osteoporose, die durch eine chronisch cholestatische Lebererkrankung hervorgerufen wird, feststellen. Wir untersuchten die Hypothese, dass bei chronisch cholestatischen Lebererkrankungen Fibronektin-Isoformen aus den aktivierten Sternzellen der Leber in die Blutbahn gelangen und dadurch die Knochenbildenden Zellen (Osteoblasten) beeinträchtigen. Es stellte sich tatsächlich heraus, dass Patienten höhere zirkulierende Isoformwerte in der Blutbahn aufwiesen als gesunde Probanden, und dass die onkofötale Fibronektin-Isoform negativ mit Osteokalzin, einem Marker der Knochenbildung, korreliert. Einen kausalen Zusammenhang bestätigten in vitro und in vivo Experimente. So führte z.B. die Injektion der Isoform in Mäuse dazu, dass die Knochenbildung gehemmt wurde und es zu Knochenverlust kam (Kawelke et al. https://www.biochem.mpg.de/en/rg/nakchbandi/publications).
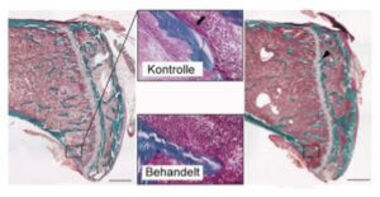
Weitere Arbeiten weisen darauf hin, dass eine O-Glykosilierung in der variablen Region des Fibronektins dafür verantwortlich ist. Wir haben den beteiligten Rezeptor identifiziert und diesen entsprechend unserer neuen Erkenntnisse modifiziert, um die Knochenbildung positiv zu beeinflussen (Sens et al. Link: http://onlinelibrary.wiley.com/doi/10.1002/jbmr.2916/abstract). Zuletzt haben wir die Funktion verschiedener Fibronektinisoformen bei der Knochenbildung charakterisiert (Bentmann et al. Link: http://onlinelibrary.wiley.com/doi/10.1359/jbmr.091011/abstract; Sens et al. Link: http://www.jbc.org/content/292/19/7745.long) und suchen nur nach Möglichkeiten uns diese neue Erkenntnisse zunutze zu machen um die Osteoporose und verschiedenen Knochenkrankheiten zu behandeln.
Skelettentwicklung
In Zusammenarbeit mit M. Moser am MPI für Biochemie werden die Effekte verschiedener Modifikationen im Integrin-Signaling Pathway in den knochenbildenden Zellen, den Osteoblasten, auf die Knochen untersucht. So führte die Ausschaltung des b1-Integrins in den Osteoblasten zu Knochenveränderungen (Phillips et al. https://www.biochem.mpg.de/en/rg/nakchbandi). Darüber hinaus untersuchen wir die Rolle der Kindlin-Moleküle und verschiedener GTPasen in verschiedenen Zelltypen des Knochens (Schmidt et al. Link: http://jcb.rupress.org/content/192/5/883.long).