
β-Thalassämie
Klinisches Beratungszentrum für Anämien Pädiatrische Onkologie, Hämatologie und Immunologie ThalassämienDefinition der Erkrankung
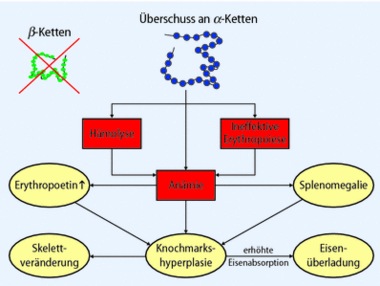
Bei der β-Thalassämie ist das β-Globingen meist durch eine Punktmutation inaktiviert, so dass die Synthese der β-Globinketten vermindert ist oder ganz fehlt. Die im Überschuss vorhandenen α-Globinketten sind schlecht wasserlöslich und präzipizieren bereits in den erythroiden Vorläuferzellen im Knochenmark. Der Überschuss an α-Globinketten führt bei homozygoten Patienten zur ineffektiven Erythropoese mit intramedullärer Hämolyse und zur transfusionsbedürftigen Anämie.
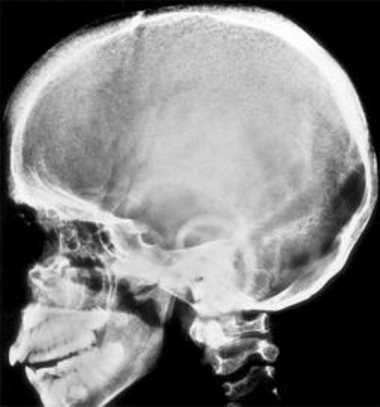
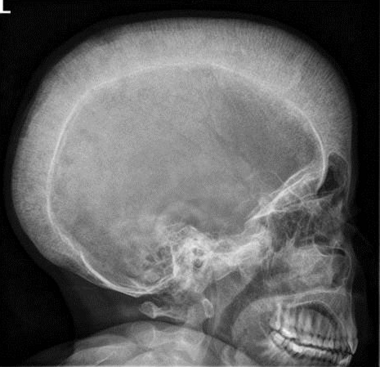
Folge der Anämie ist eine erheblich gesteigerte Erythropoese, die zur erythroiden Hyperplasie des Knochenmarks und zu Skelettdeformitäten (Facies thalassaemica und Bürstenschädel) sowie zum erhöhten Folat-und Energiebedarf führt. In der Regel besteht lebenslanger und regelmäßiger Transfusionsbedarf. Durch die Transfusionen aber auch durch eine erhöhte intestinale Eisenresorption entsteht eine Hämosiderose, mit ihren Komplikationen Kardiomyopathie, Leberfibrose, Leberzirrhose sowie multiplen endokrinen Ausfällen.
Symptome
Klinisches Bild, Verlaufsformen und Diagnosestellung
Thalassaemia major
Die Diagnose einer Thalassemia major (homozygote β-Thalassämie) wird meist im 2. Lebenshalbjahr gestellt. Erste klinische Symptome sind Gedeihstörung, zunehmende Blässe, Infektneigung oder aufgetriebenes Abdomen infolge Hepatosplenomegalie.
Bei unzureichender Behandlung kommt es zu Wachstumsretardierung, Skelettanomalien, Kachexie und massiver Hepatosplenomegalie. Bleibt eine adäquate Therapie weiterhin aus, stehen die Folgen der zunehmenden Anämie und der dadurch bedingten erythroiden Hyperplasie des Knochenmarks im Vordergrund.
Der weitere Verlauf führt zu rezidivierenden Infektionen, pathologischen Frakturen, Hypersplenismus, Cholelithiasis und zu einer Vielfalt von Verdrängungssyndromen aufgrund der extramedullären Erythropoese. In ihrem natürlichenVerlauf versterben Patienten mit einer Thalassaemia major noch im Kleinkindalter an den Folgen der Anämie.
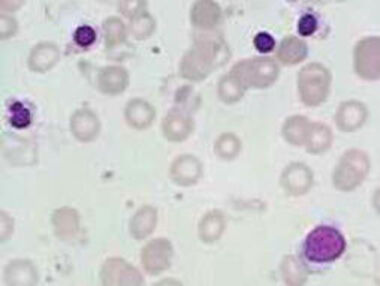
Das Blutbild zeigt immer eine schwere Anämie mit Anisozytose und Poikilozytose. Im Blutausstrich finden sich Targetzellen und Erythroblasten
Gesichert wird die Diagnose durch eine Hb-Elektophorese, die einen deutlich erhöhten Anteil von HbF aufweist.
Thalassaemia minor
Die Thalassaemia minor ist die heterozygote Form der Thalassämie, sie weist keine relevanten klinischen Veränderungen auf. Im Blutbild findet sich keine oder eine nur geringgradige und keinesfalls transfusionspflichtige mikroytäre hypochrome Anämie. Diagnostisch entscheidend sind hier normale Eisenspeicher bei erhöhtem HbA2, die die Unterscheidung zur Eisenmangelänamie ermöglichen und Betroffene vor unnötigen Eisensubstitutionen bewahren.
Cave: Bei heterozygoter Thalassämie und gleichzeitig vorliegendem Eisenmangel kann die HbA2-Erhöhung fehlen. Ist der Eisenmangel behoben und es besteht weiterhin eine Mikrozytose, empfiehlt sich die Wiederholung der Hb-Elektrophorese.
Die einzige mögliche Komplikation ist eine stärker als gewöhnlich ausgeprägte Schwangerschaftsanämie.
Wichtig ist bei der Diagnose einer heterozygoten β-Thalassämie auch die Untersuchung beider Eltern, um ein ggf. bestehendes humangenetisches Risiko für die homozygote Form zu identifizieren und den Eltern eine spezifische humangenetische Beratung zu ermöglichen.
Thalassaemia intermedia
Zum klinisch milderen Bild der Thalassaemia intermedia, d.h. zu einer nicht regelmäßig oder gar nicht transfusionsbedürftigen Anämie kann es allerdings auch bei der homozygoten Form der β-Thalassämie kommen, wenn der α-Globinkettenüberschuss weniger stark ausgeprägt ist [10]. Dies kann durch eine hohe Restaktivität der β-Globingene, durch eine gleichzeitig vorliegende α-Thalassämie oder durch genetische Veränderungen des β-Globingenkomplexes zustande kommen, die zu einer postnatal persistierenden γ-Globingenexpression und somit zur hereditären Persistenz der fetalen Hämoglobinsynthese (HPFH) führen (s.Tabelle 2) [9, 11].
Pathogenese der Thalassaemia intermedia
Homozygote b-Thalassämie
- Thalassämiemutationen mit einer hohen Restaktivität des b-Globingens
- HPFH-Mutationen
- a-Thalassämiedeletionen
Heterozygote b-Thalassämie
- triplizierte a-Globingene
- autosomal dominante b-Thalassämie
- instabile b-Globinketten
a-Thalassämie
- HbH-Krankheit
Diagnose
Ablauf der Behandlung
Schwerpunkte der Therapie
Knochenmarktransplantation
Die einzige kurative Behandlung ist die Knochenmarktransplantation.
Weltweit wurden bei Patienten mit Thalassaemia major mehr als 1000 Knochenmarktransplantationen erfolgreich mit verwandten, HLA-identischen Spendern durchgeführt [8].
Die Komplikationen der Stammzelltransplantation sind bei jungen Patienten ohne Hämosiderose und konsekutiver Leberfibrose gering, wenn ein HLA-identisches Geschwister als Stammzellspender zur Verfügung steht [12]. Daher sollte die Transplantation möglichst vor dem Schulalter durchgeführt werden.
Bluttransfusionen
Seit den 60er Jahren gehören regelmäßige Bluttransfusionen zum Standard der konventionellen Therapie.
Die Ziel der Transfusionen beinhaltet die Korrektur der Anämie und somit die Suppression der gesteigerten Erythropoese.
Kontrolle: Retikulozyten < 5%o weisen auf eine ausreichende Hemmung der gesteigerten Erythropoese hin
Adäquate Transfusionen ermöglichen ein normales Wachstum und Entwicklung und erlauben eine normale Belastungsfähigkeit. Sie verhindern Komplikationen wie Herzinsuffizienz und schwere Knochendeformitäten.
Die Bluttransfusionen sind lebenslang notwendig, bis eine kurative Behandlung wie die allogene Knochenmarktransplantation durchgeführt wird.
Die Indikation für die regelmäßige Transfusion wird bei einem steady state Hämoglobinwert von < 7-8 g/dl gestellt.
Der Basis-Hb (Hb-Wert vor Transfusion) sollte 9 g/dl in den transfusionsfreien Intervallen nicht unterschreiten, damit eine ausreichende Suppression der endogenen Erythropoese erreicht wird.
Transfusionsintervalle und Transfusionsmenge
Zur Reduktion des Transfusionsvolumen sollten die Abstände zwischen den Transfusionen so kurz wie möglich sein, müssen aber auch mit dem normalen Lebensablauf des Patienten vereinbar sein. Als Kompromiss werden Transfusionen in dreiwöchentlichem Abstand empfohlen.
Bei einem Transfusionsintervall von 3 Wochen und einer Überlebenszeit der transfundierten Erythrozythen von etwa 100 Tagen bedeutet dies, dass ein Hb von 12-13 g/dl nach der Transfusion erreicht werden sollte. Das dazu benötigte Transfusionsvolumen errechnet sich nach der Formel 1:
Ery-Konzentrat [ml] = (Hbsoll-Hbist [g/dl]) x KG [kg] x VkgKG[ml/kg] : HbKons[g/dl]
Hbsoll = angestrebtes Hb nach Transfusion
Hbist = Hb vor Transfusion
HbKons = Hb der Erythrozythenkonserve
KG = Körpergewicht
VkgKG = Blutvolumen /kgKG
Das Blutvolumen kann im steady state mit 80 ml/kgKG angenommen werden. Die meisten Konserven werden mit einem Hb von » 27g/dl geliefert. Formel 1 kann dann in Formel 2 vereinfacht werden:
Ery-Konzentrat [ml] = (Hbsoll-Hbist [g/dl]) x KG [kg] x 3
Zu Beginn der Behandlung kann das benötigte Blutvolumen höher liegen. Die benötigte Transfusionsmenge sollte dokumentiert werden, um den individuellen Bedarf der Patienten zu ermitteln. Bei einem steigenden Bedarf muss differentialdiagnostisch an eine Milzsequestration oder eine Immunhämolyse gedacht werden.
Chelattherapie
Der Erfolg der Konventionellen Therapie ist maßgeblich von der Eisenelimination abhängig [1, 15].
Seit ca. 40 Jahren werden Patienten mit Chelatbildnern therapiert. Trotz enormer Verbesserung der Lebenserwartung durch die Chelattherapie ist die führende Todesursache die Kardiomyopathie aufgrund myokardialer Eisenablagerung.
Alle Patienten, die regelmäßig transfundiert werden, benötigen eine Therapie mit Chelatbildnern. Da die Risiken der Nebenwirkungen dieser Medikamente bei einer niedrigen Eisenbelastung am höchsten sind, sollte abgewartet werden, bis sich ein ausreichend chelierbarer Eisenpool gebildet hat.
Die Therapie sollte bei einer Leber-Eisenkonzentration > 3 mg/g Lebertrockengewicht (s. Quantifizierung der Hämosiderose) begonnen werden. Dies ist in der Regel nach einem Jahr regelmäßiger Transfusionen der Fall. Es stehen momentan drei Medikamente zur Eisenelimination zur Verfügung, die in Tabelle 3 zusammengefaßt sind.
Deferoxamin (Desferal®)
Seit den 70er Jahren ist Deferoxamin (DFO) das Standardmedikament zur Eisenchelation. Es wirkt sicher und effektiv bei transfusionsbedingter Hämosiderose. Es bindet Eisen, und der Eisen-DFO-Komplex wird über Stuhl und Urin ausgeschieden.
Die Applikation des DFO kann nur parenteral erfolgen, da DFO nicht resorbiert wird. Außerdem ist seine Halbwertszeit sehr kurz, so dass die Wirksamkeit von der Länge der DFO- Infusion abhängt. Pharmakokinetisch ideal wäre daher eine 24-stündige Infusion an 7 Tagen die Woche. Da dies in der Regel nicht praktikabel ist, hat sich eine Applikation über Nacht als Standardtherapie etabliert. Die gelegentlich empfohlene Infusion an nur 5 Tagen der Woche reicht für eine ausgeglichene Eisenbilanz nicht aus und ist daher nicht zu empfehlen. Auch die nächtliche Infusion sollte über so viele Stunden wie möglich erfolgen; bei vielen Patienten lässt sich eine 12-stündige Infusionsdauer ohne zusätzliche Einschränkung der Lebensqualität realisieren.
Ist die Indikation zur Therapie mit Deferoxamin gestellt, sollten folgende Dosierungen verwendet werden:
- Start bei Leber-Eisenkonzentration > 3 mg/g Lebertrockengewicht:
25 mg/kg KG 5 mal pro Woche (nach ca. 1 Jahr regelmäßiger Transfusion) - Leber-Eisenkonzentration 7-15 mg/g:
40 mg/kg KG 7 mal pro Woche (bei Serumferritin-Werten von 1000 ug/l) - Leber-Eisenkonzentration > 15 mg/g:
50 mg/kg KG 7 mal pro Woche möglichst als (kontinuierliche Infusion)
DEFERIPRON (FERRIPROX®)
Deferipron ist ein oral wirksamer Chelator der 1987 zum ersten Mal bei Menschen angewendet wurde. Heute ist Deferipron in Deutschland bei Patienten zugelassen, bei denen eine Intoleranz gegenüber Deferoxamin besteht. Deferipron führt zu einer Eisenelimination über den Urin und wird in einer Dosis von 3 x 25 mg/kgKG eingesetzt.
Deferipron verursacht häufig gastrointestinale Nebenwirkungen, seltene schwere Nebenwirkungen sind erosive Arthritis, Neutropenie, bis hin zur schweren Agranulozytose, die aber in der Regel transitorisch sind [5]. Aufgrund der relevanten Nebenwirkungen wird Deferipron vor allem bei Patienten mit schwerer Eisenüberladung in Kombination mit Deferoxamin eingesetzt, um kardialen Komplikationen der Siderose entgegen zu wirken.
DEFERASIROX (EXJADE®)
Deferasirox ist ein oraler Eisenchelator, der in Deutschland seit 2006 für die Eiseneliminationstherapie bei Kindern ab 2 Jahren zugelassen ist.
Die Effizienz von Deferasirox wurde in einer großen, internationalen, multizentrischen randomisierten Studie gezeigt [2]. Deferasirox führte in dieser Studie zu einer dosis-abhängigen Reduktion des Serum- Ferritins und parallel dazu zu einer Abnahme der Lebereisenkonzentration. Mit einer Plasma-Halbwertszeit von 8 bis 16 Stunden ist eine einmalige orale Dosis ausreichend. Die Deferasirox-Eisen-Komplexe werden mit dem Stuhl ausgeschieden [7], [13].
Mit Desferasirox steht ein neues Medikament für die Eiseneliminationstherapie zur Verfügung. Obgleich mit Deferoxamin bereits ein gut etabliertes, wirksames Medikament vorhanden ist [6], treten Hämosiderose bedingte Komplikationen immer noch bei vielen Patienten auf, da der Applikationsmodus aufwendig, und die Compliance daher häufig schlecht ist. Solange jedoch keine Langzeit-Daten zu Deferasirox, insbesondere zu Nebenwirkungen wie Leber- und Nierenfunktion sowie zum Einfluss auf Wachstum und Entwicklung vorliegen, ist nach wie vor Deferoxamin als Medikament der Wahl zu empfehlen. Weitere klinische Studien sind notwendig, um Deferoxamin als Goldstandard abzulösen. Bei Patienten mit schlechter Compliance stellt Deferasirox eine Alternative dar.
Vergleich der verfügbaren Eisenchelatoren
| Deferoxamin | Deferasirox | Deferipron | |
Anwendung | parenteral, meist subcutan o. intravenös | Oral | Oral |
Plasma-HWZ | kurz (min), benötigt kontante Zufuhr | lang, 8-16 h, bleibt 24h im Plasma | < 2h, Einnahme mindestens 3x/d |
Molare Effizienz Eisen zu binden | Hoch | moderat | niedrig |
Wichtige NW | oto-u.okulotoxisch, lokale Hautreaktionen an der Injektionsstelle, in hohen Dosen: Knochen-u.Gelenkschmerzen, Beeinträchtigung des Wachstums, Ventilationsstörung der Lunge in hohen Dosierungen | abdominelle Beschwerden, Hautausschlag o. milde Diarrhö bei Therapiebeginn, milder Anstieg des Kreatinins | gastrointestinale Nebenwirkungen, transitorische Blutbild- veränderungen (Neutropenie bis Agranulozytose), erosive Arthritis |
Fähigkeit intrazelluläres kardiales Eisen u. Eisen in anderen Geweben zu binden | wahrscheinlich geringer als Deferipron u. Deferasirox | bisher noch nicht genügend klin. Daten vorhanden, vielversprechend in in vitro Studien | hoch in klin. und in vitro Studien |
Quantifizierung der Hämosiderose
Um den geeigneten Beginn der Eiseneliminationstherapie zu bestimmen sowie zur Früherkennung der durch die Transfusionen bedingten Nebenwirkungen, ist die Abschätzung der Eisenüberladung von großern Bedeutung. Das Serumferritin ist erheblichen Tagesschwankungen unterlegen und kann nur begrenzt zur Verlaufskontrolle verwendet werden. Der empfindlichste Parameter für die Diagnose einer Eisenüberladung ist die Transferrinbindungskapazität. Sie erreicht jedoch schon sehr früh ein Plateau und ist daher für die Verlaufskontrolle von Patienten mit einer Thalassaemia major nicht geeignet. Die Leberbiopsie ermöglicht die direkte Messung derEisenbelastung, sie stellt jedoch als invasives Verfahren eine Belastung des Patienten dar und ist somit nur in besonderen Situationen angemessen. Nicht-invasiv kann die Lebereisenkonzentration mit einem superconducting quantum interference device (SQUID) gemessen werden. Diese Messung korreliert in exzellenter Weise mit der direkt gemessenen Lebereisenkonzentration und ist für die Steuerung der Eiseneliminationstherapie somit ideal geeignet. Der technische Aufwand dieser Messmethode ist jedoch erheblich, so dass das SQUID weltweit nur an vier Zentren [13] in Deutschland aktuell nur an der Universitätsklinik Hamburg-Eppendorf verfügbar ist. Als Alternative zur nicht-invasiven Lebereisenmessung hat sich in den vergangenen Jahren die Kernspintomographie etabliert. Mit Hilfe spezieller Sequenzen (T2*) kann nicht nur die Lebereisenbelastung, sondern auch die Herzeisenbelastung quantifiziert werden [16].
Literaturhinweise
- Aessopos A, Farmakis D, Hatziliami A, Fragodimitri C, Karabatsos F, Joussef J, Mitilineou E, Diamanti-Kandaraki E, Meletis J and Karagiorga M (2004) Cardiac status in well-treated patients with thalassemia major. Eur J Haematol 73: 359-66
- Cappellini MD, Cohen A, Piga A, Bejaoui M, Perrotta S, Agaoglu L, Aydinok Y, Kattamis A, Kilinc Y, Porter J, Capra M, Galanello R, Fattoum S, Drelichman G, Magnano C, Verissimo M, Athanassiou-Metaxa M, Giardina P, Kourakli-Symeonidis A, Janka-Schaub G, Coates T, Vermylen C, Olivieri N, Thuret I, Opitz H, Ressayre-Djaffer C, Marks P and Alberti D (2006) A phase 3 study of deferasirox (ICL670), a once-daily oral iron chelator, in patients with beta-thalassemia. Blood 107: 3455-62
- Chui DH (2005) Alpha-thalassemia: Hb H disease and Hb Barts hydrops fetalis. Ann N Y Acad Sci 1054: 25-32
- Cohen AR, Galanello R, Pennell DJ, Cunningham MJ and Vichinsky E (2004) Thalassemia. Hematology Am Soc Hematol Educ Program 14-34
- Cohen AR, Galanello R, Piga A, De Sanctis V and Tricta F (2003) Safety and effectiveness of long-term therapy with the oral iron chelator deferiprone. Blood 102: 1583-7
- Gabutti V and Piga A (1996) Results of long-term iron-chelating therapy. Acta Haematol 95: 26-36
- Galanello R (2005) Evaluation of ICL670, a once-daily oral iron chelator in a phase III clinical trial of beta-thalassemia patients with transfusional iron overload. Ann N Y Acad Sci 1054: 183-5
- Giardini C (1997) Treatment of beta-thalassemia. Curr Opin Hematol 4: 79-87
- Kulozik AE (1992) Beta-thalassaemia: molecular pathogenesis and clinical variability. Eur J Pediatr 151: 78-84
- Kulozik AE (2005) Thalassämien. In: Gadner H, Gaedike G, Niemeyer C and Ritter J (Hrsg) Springer Verlag, Heidelberg, 169-178
- Kulozik AE, Bellan-Koch A, Kohne E and Kleihauer E (1992) A deletion/inversion rearrangement of the beta-globin gene cluster in a Turkish family with delta beta zero-thalassemia intermedia. Blood 79: 2455-9
- Lucarelli G, Giardini C and Angelucci E (1997) Bone marrow transplantation in thalassemia. Cancer Treat Res 77: 305-15
- Neufeld EJ (2006) Oral chelators deferasirox and deferiprone for transfusional iron overload in thalassemia major: new data, new questions. Blood 107: 3436-41
- Singer ST, Styles L, Bojanowski J, Quirolo K, Foote D and Vichinsky EP (2000) Changing outcome of homozygous alpha-thalassemia: cautious optimism. J Pediatr Hematol Oncol 22: 539-42
- Wolfe L, Olivieri N, Sallan D, Colan S, Rose V, Propper R, Freedman MH and Nathan DG (1985) Prevention of cardiac disease by subcutaneous deferoxamine in patients with thalassemia major. N Engl J Med 312: 1600-3
- Gandon Y, Olivie D, Guyader D, Aube C, Oberti F, Sebille V, et al. Non-invasive assessment of hepatic iron stores by MRI. Lancet 2004; 363:357-6
Mögliche Komplikationen / Risiken
Therapie der Komplikationen
Die beste Therapie ist die Prävention der Hämosiderose durch eine konsequent durchgeführte Eiseneliminationstherapie Die Komplikationen entwickeln sich langfristig aufgrund der Hämosiderose, die durch die regelmäßigen, lebensnotwendigen Transfusionen entsteht. Die bedeutendste, die Mortlität der Erkrankung bestimmende Komplikation ist die Kardiomyopathie. Weitere Todesursachen sind Infektionen und eine chronische Lebererkrankung. Die Therapie der Kardiomyopathie erfolgt symptomatisch mit Digitalispräparaten und Antiarrhythmika. Ein kausaler Ansatz ist die kontinuierlich i.v. durchgeführte DFO-Therapie, die die Eisenüberladung vermindern und somit die Herzfunktion verbessern kann. Bei endokrinen Komplikationen wie Kleinwuchs und Pubertas tarda, Hypothyreose und Hypoparathyreoidismus empfiehlt sich die entsprechende Hormonsubstitution. Ein Diabetes mellitus tritt v.a. bei Patienten mit schwerer Hämosiderose und Leberfunktionsstörungen auf. Ist die Glukosetoleranz gestört, manifestiert sich meist nur wenige Monate später ein Diabetes mellitus, der zunächst mit oralen Antidiabetika, bald aber auch mit Insulin behandelt werden muss.