
Thalassämien
Klinisches Beratungszentrum für Anämien Pädiatrische Onkologie, Hämatologie und ImmunologieDefinition der Erkrankung
Thalassämien sind eine Gruppe von autosomal und bis auf wenige Ausnahmen rezessiv vererbte Erkrankungen, bei denen die Synthese einer oder mehrerer Globinketten beeinträchtigt ist. Pathogenetisch liegt eine Mutation der Globingene bzw. deren regulatorischer DNA-Sequenzen vor. Endemisch kommen die Thalassämien (griech. „thalassa“ = Meer) nicht nur im Mittelmeerraum, sondern auch in weiten Teilen Asiens und in Afrika vor. Durch die Einwanderung von Menschen aus diesen Teilen der Welt gehören die Thalassämien auch bei uns zu den häufigen Erkrankungen in der pädiatrischen Hämatologie. Insgesamt werden an deutschen Kinderkliniken derzeit etwa 400-500 Patienten mit einer transfusionsbedürftigen homozygoten β-Thalassämie behandelt.
Die größte klinische Bedeutung haben bei uns die β-Thalassämien; α-Thalassämien und andere Thalassämieformen/-syndrome sind in Deutschland viel seltener.
Bei den Thalassämien ist die Synthese der Polypeptidketten gestört. Nach der betroffenen Polypeptidkette spricht man von α- oder β-Thalasämie.
Die β-Thalassämien haben in Europa die größte klinische Bedeutung. Die Erhöhung des Anteils an HbF am Gesamt-Hb wird vornehmlich durch die stark verminderte oder fehlende β-Globinkettensynthese verursacht. Insofern ist das prozentual erhöhte HbF bei der homozygoten β-Thalassämie kein Ausdruck eines Kompensationsmechanismus. Hinzu kommt jedoch erstens eine Selektion der auch physiologischerweise vorkommenden kleinen Population von HbF Zellen, da diese Zellen (nicht so stark) vom α-Globinkettenüberschuss betroffen sind. Zweitens wird diese kleine Zellpopulation wegen der allgemeinen Expansion des Erythroiden Systems auch vermehrt, so dass hier ein gewisser, bei der Thalassaemia major jedoch keineswegs ausreichender Kompensationsprozess vorhanden ist.
α-Thalassämien und andere Thalassämieformen/-syndrome sind viel seltener. Bei α-Thalassämien entstehen vermehrt β- bzw. γ-Tetramere; β-Tetramere werden als HbH, γ-Tetramere als HbB oder HbBarts bezeichnet. Diese pathologischen Überschusshämoglobine sind diagnostisch für den Hb Bart’s Hydrops fetalis bzw. für die HbH Krankheit.
Symptome
Diagnose
Struktur und Funktion des Hämoglobins
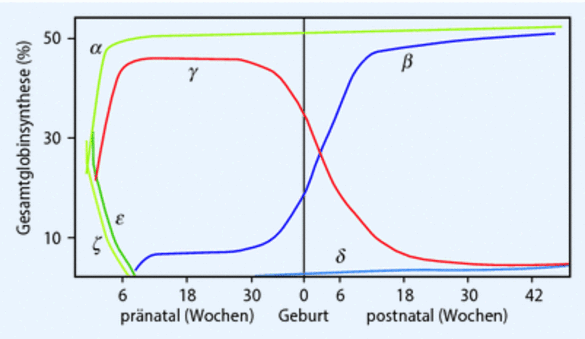
Das Hämoglobinmolekül besteht aus vier Häm-Gruppen, mit einem Protoporphyrin-Ring als eisentragendem Grundgerüst und aus vier Globin-Polypeptidketten, von denen je zwei identisch sind. Im Laufe der Entwicklung des menschlichen Organismus werden unterschiedliche Hämoglobine gebildet. Ungefähr 14 Tage nach der Konzeption werden im Dottersack die Hämoglobine Gower I (ζ2ε2)und II (α2ε2) und Hb Portland (ζ2γ2) sowie wenig später das fetale Hämoglobin HbF (α2γ2) synthetisiert. Nach ca. 8 Wochen werden die embryonalen Gene inaktiviert und HbF wird das vorherrschende Hämoglobin der Intrauterinperiode. Das adulte HbA wird in dieser Phase nur in kleinen Mengen gebildet. Etwa zum Geburtstermin wird die γ- durch die β- Globinsynthese abgelöst, das fetale Hämoglobin HbF fällt exponentiell und parallel zur Steigerung der adulten Hämoglobinsynthese ab (Abbildung1). Die prozentuale Normverteilung (Mittelwerte von elektrophoretischen Messungen) ist in Tabelle 1 aufgeführt.
Hb-Verteilung bei Normalpersonen und bei Thalassämie
Hämoglobin | Ketten | normal | erhöht bei | vermindert bei |
HbA1 | α2β2 | ca. 97 % | homozygoter | |
HbA2 | α2δ2 | 2-3,4 % | heterozyote b-Thalassämie ( T. minor): 3,5-6 % | heterozygoter α0-Thalassämie |
HbF | α2γ2 | 0,5 % Neugeborene: >70 % | b-Thalassaemia major: > 90 % b-Thalassaemia minor: variabel bis zu 5 % Thalassaemia intermedia: variabel | |
HbH | β4 | 0 % | HbH-Krankheit | |
HbBarts | γ4 | 0 % | Hb Bart’s Hydrops fetalis |
Ablauf der Behandlung
Patienten mit schweren Formen der Thalassämien benötigen eine komplexe konventionelle Therapie, die aus Transfusionen, Eiseneliminationen und supportiven Maßnahmen besteht. Steht ein Stammzellspender zur Verfügung, ist die frühe Transplantation, möglichst noch vor dem Schulalter, die Behandlungsmethode der Wahl.