




Das Long QT-Syndrom im Kindes- und Jugendalter
Definition des Long QT-Syndroms (LQTS)
Das Long QT-Syndrom umfasst eine Gruppe von seltenen, genetisch bedingten, funktionellen Störungen verschiedener Ionenkanäle der Zellmembran des Herzens. Diese Störungen gehen mit einem verlängerten QT-Intervall im Elektrokardiogramm (EKG), einer veränderten elektrischen Repolarisation der Herzkammern sowie dem Auftreten bestimmter Herzrhythmusstörungen (ventrikuläre Tachykardien in Form von Torsade de Pointes) und einer hohen Prädisposition für kardiale Ereignisse einher.
Historisches
Jervell und Lange-Nielsen berichteten im Jahre 1957 neben Romano 1963 und Ward 1964 erstmals über die klinischen Manifestationen und die beobachteten Erbgänge des Long QT-Syndroms, wobei dieses Krankheitsbild bereits zuvor, nicht wissentlich seiner kardialen Ursache, in einzelnen Fallbeschreibungen Erwähnung fand.
Der entscheidende Durchbruch für das Verständnis der klinischen Manifestation des Long QT-Syndroms bzw. der zugrunde liegenden gemeinsamen Mechanismen erfolgte mit der Entdeckung einer genetisch bedingten Störung als Ursache des Long QT-Syndroms. Mit Hilfe von Klonierungstechniken konnten Keating et al. 1991 bei verschiedenen Familien erstmalig eine Beziehung zum kurzen Arm des Chromosoms 11 herstellen. Biophysikalische Untersuchungen der codierten Aminosäuresequenzen ergaben, dass es sich um Natrium- und Kalium-Ionenkanäle handelte, die insbesondere in der Herzmuskulatur vorkommen und bei Depolarisations- und Repolarisationsvorgängen an der Zellmembran während des Ablaufs des Aktionspotentials eine wichtige Funktion ausüben.
Formen des Long QT-Syndroms
Das Long QT-Syndrom kann in familiären, sporadischen und erworbenen Formen vorkommen. Die Prävalenz des Long QT-Syndroms liegt nach neueren Untersuchungen bei bis zu 1 auf 2.000 Lebendgeborene.
Familäre Formen des Long QT-Syndrom
Zur familiären Form gehört das bereits erwähnte Jervell/Lange-Nielsen-Syndrom, das autosomal rezessiv vererbt wird und in den meisten Fällen mit einer angeborenen Schwerhörigkeit einhergeht. Das häufigere Romano/Ward-Syndroms folgt einem autosomal dominantem Erbgang und zählt ebenfalls zur familiären Form. Beide lassen sich aufgrund verschiedener veränderter Gene in weitere Unterformen (Subtypen) unterteilen.
Sporadische Formen des Long QT-Syndroms
Zu den sporadischen Fällen des Long QT-Syndroms werden jene Patienten gezählt, die innerhalb ihrer Familie der erste entdeckte Fall sind (Indexpatient), wobei sich die übrigen Familienmitglieder klinisch und genetisch als unauffällig erweisen müssen, während der Betroffene sowohl symptomatisch als auch genetisch identifizierbar sein muss.
Erworbene Form des Long QT-Syndrom
Unter einem erworbenen Long QT-Syndrom wird ein Funktionszustand der Herzmuskelzellen verstanden, bei dem vorübergehend, ausgelöst durch äußere Einflüsse, die Aktivität der myokardialen Ionenkanäle funktionell dem angeborenen Long QT-Syndrom entspricht, jedoch bei Entfernung der exogenen Einflüsse wieder in den Normalzustand zurückkehrt. Eine erworbene QT-Verlängerung kann beispielsweise unter der Einnahme bestimmter Medikamenten und bei Veränderungen der Elektrolyte im Blut vorkommen.
Klinisches Erscheinungsbild des Long QT-Syndroms
Vorgeburtlich kann sich ein Long QT-Syndrom bereits beim Fetus in Form einer erniedrigten Herzfrequenz manifestieren.
Zu kardialen Ereignissen (Herzrhythmusstörungen, Präsynkopen, Synkopen, Herzstillstand) im Rahmen eines Long QT-Syndroms kann es durch körperlichen und emotionalen Stress kommen, ausgelöst beispielsweise durch Sport, Furcht, Zorn, Freude, Aufregung und plötzliches Wecker-/Telefonläuten. Auch beim Springen ins Wasser bzw. Tauchen sind kardiale Ereignisse möglich.
Bei einzelnen Patienten sind auch in Ruhephasen und während des Schlafes kardiale Ereignisse aufgetreten.
Elektrokardiographische Veränderungen beim Long QT-Syndrom
Die Störungen der elektrischen Repolarisation der Herzmuskelzellen beim Long QT-Syndrom zeigen sich in bestimmten Veränderungen im Oberflächen-EKG. Definitionsgemäß zählt hierzu ein verlängertes QT-Intervall (Abb.1).
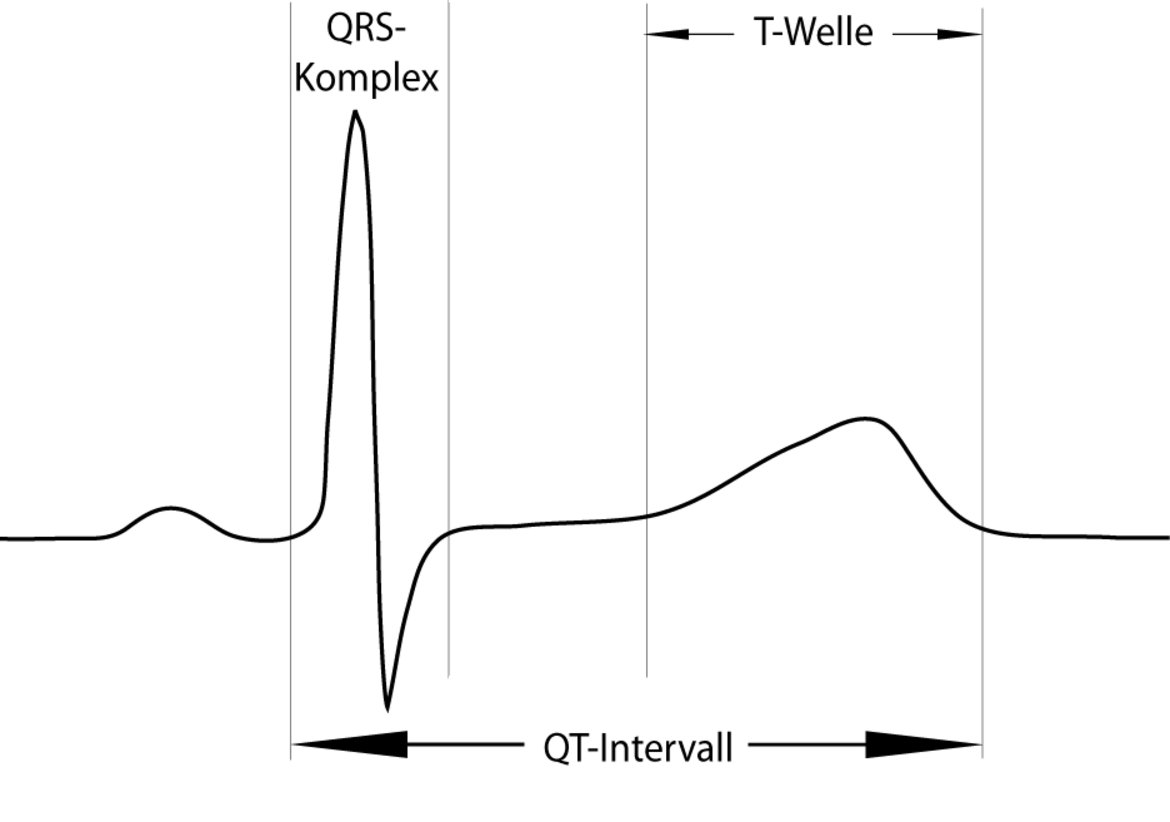
Dabei muss berücksichtigt werden, dass das QT-Intervall u.a. abhänigig ist von Herzfrequenz, Geschlecht sowie Alter des Patienten. Im klinischen Alltag wird daher der frequenzkorrigierte QTc-Wert, üblicherweise nach der Formel von Bazett, verwendet:
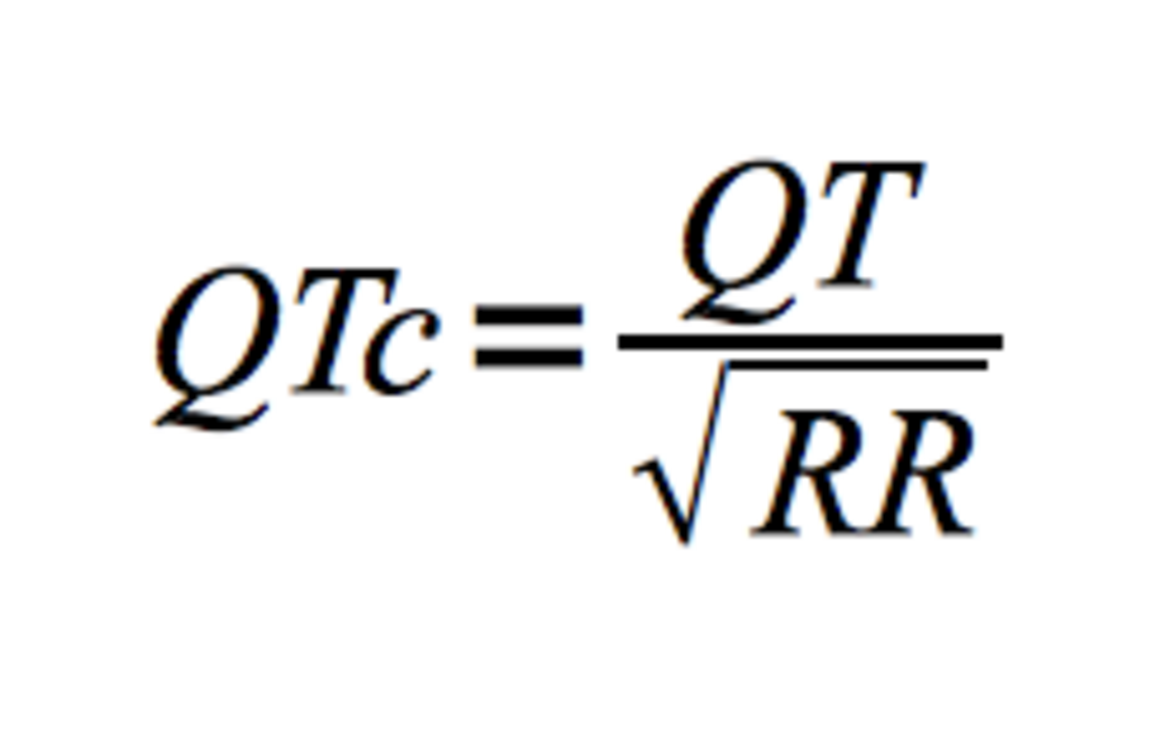
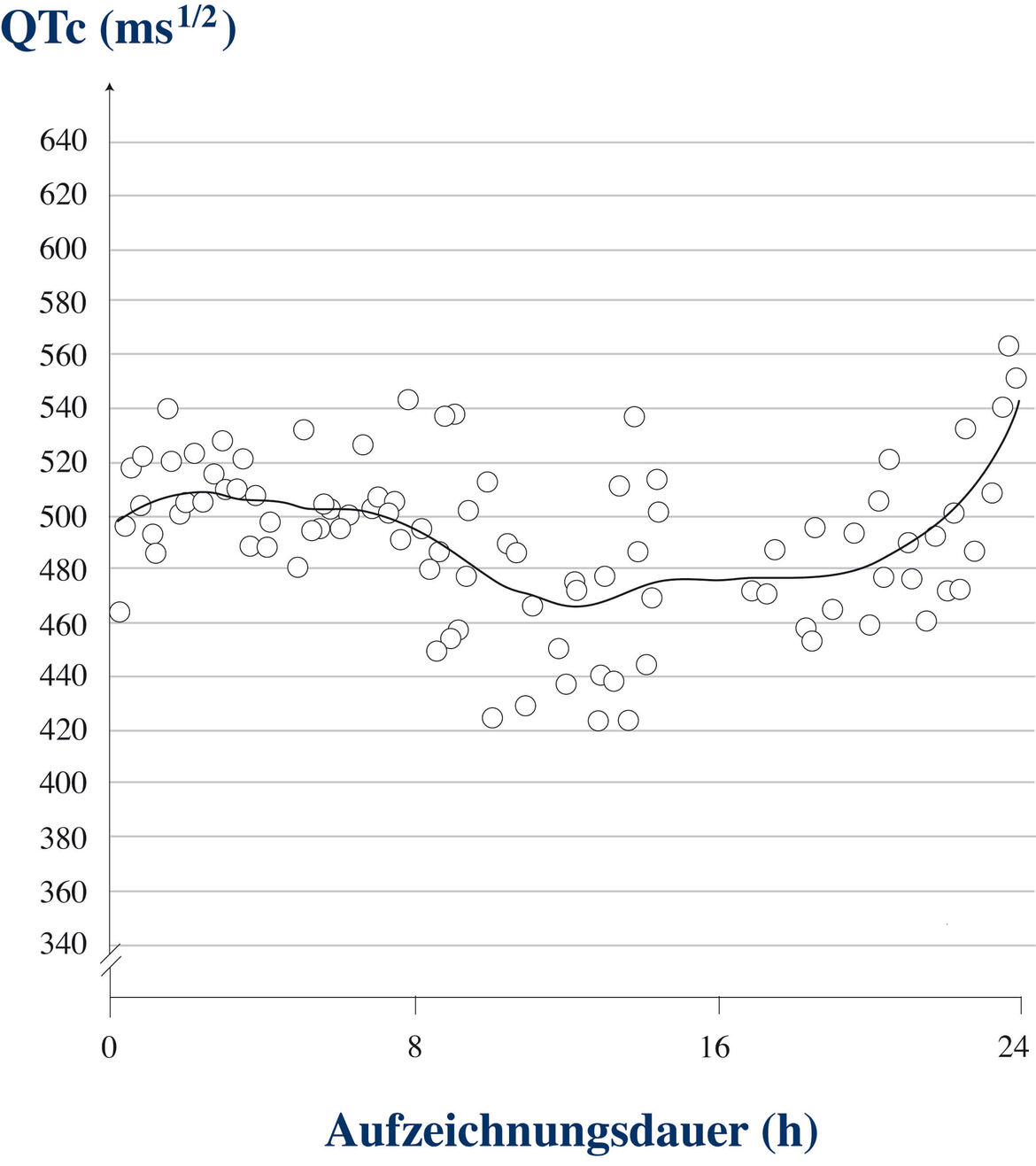
Diese QTc-Werte zeigen im Tagesverlauf oft eine deutliche Variation (Abb.2):
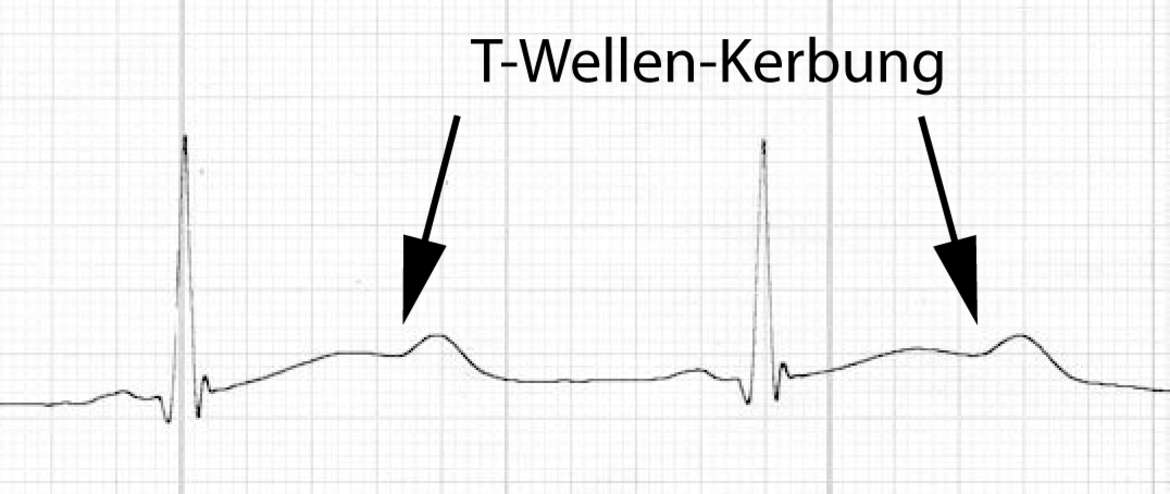
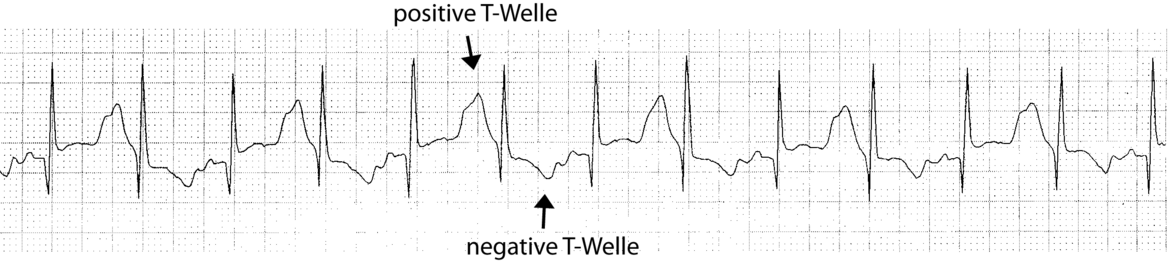
Desweiteren finden sich beim LQTS auch charakteristische morphologische Veränderungen der T-Welle wie Kerbungen am auf- oder absteigenden Teil der T-Welle (Abb.3).
Eine weitere T-Wellen-Veränderung stellt der makroskopische T-Wellen-Alternans dar (Abb.4). Er bezeichnet im EKG eine sich von Schlag zu Schlag alternierend verändernde T-Wellen-Morphologie hinsichtlich T-Wellen-Amplitude bzw. T-Wellen-Ausrichtung. Im engeren Sinne wird der Begriff des T-Wellen-Alternans für eine sich von Schlag zu Schlag ändernde T-Wellen-Ausrichtung verwendet.
Diagnosestellung des Long QT-Syndroms im Kindesalter
Im typischen Fall mit rezidivierenden, stressinduzierten Synkopen bei nachweislich deutlich verlängertem QT-Intervall im EKG und gegebenenfalls familiärem Vorliegen einer entsprechenden Symptomatik stellt die Diagnosestellung eines Long QT-Syndroms im Kindesalter kein Problem dar.
Über lange Zeit war die Verlängerung des QT-Intervalls bzw. des QTc-Wertes, d.h. der frequenzkorrigierten QT-Zeit, im Standard-EKG traditionell das einzige und absolute Kriterium für die Diagnosestellung eines Long QT-Syndroms in einem klinisch hierfür verdächtigen Fall.
Als eines der Ergebnisse der Auswertung einer 1979 initiierten Internationalen LQTS-Registry wurde 1985 eine erste Version des Schwartz-Scores formuliert und zuletzt in 2011 aktualisiert (Abb.5). Die klinische Diagnosestellung eines LQTS wird heute bei Erwachsenen als auch bei Kindern und Jugendlichen mit Hilfe dieser inzwischen allgemein anerkannten Kriterien nach Schwartz gestellt. Danach gehen neben der Dauer des QT-Intervalls u.a. T-Wellen-Veränderungen, klinische Symptome und eine eventuelle familiäre Belastung in einen Punktescore ein. Die hier ermittelte Gesamtpunktezahl gibt einen Anhalt für die Wahrscheinlichkeit des Vorliegens eines Long QT-Syndroms.

Seit der ersten Identifizierung eines Mutationspunktes 1991 durch Keating et al. sind inzwischen mehrere Genotypen gefunden worden. Die Diagnose kann somit heute molekulargenetisch gesichert und nach den jeweiligen Genotyp klassifiziert werden.
Therapeutische Möglichkeiten beim LQTS im Kindes- und Jugendalter
Gegenwärtig ist eine kausale Therapie des Long QT-Syndroms nicht möglich. Umso wichtiger ist daher die Vorbeugung vor möglichen Rhythmusstörungen (Torsade de Pointes), die zu Prä- oder Synkopen, aber auch zum plötzlichen Herztod führen können. Mit der Durchführung eines angemessenen Behandlungskonzeptes kann zwar das Risiko gesenkt, aber weiterhin noch nicht vollständig beseitigt werden.
Allgemeine Maßnahmen
Zu den Behandlungsstrategien gehören allgemeine sowie spezielle Maßnahmen. Erstgenannte beinhalten die Vermeidung triggernder Auslösemechanismen. Hierzu zählen z.B. das Unterlassen sportlicher Aktivitäten unter Leistungsdruck (z.B. Schulsport), insbesondere Wassersport und Tauchen. Vermieden werden sollten ferner psychische Stresssituationen (z.B. Weckerläuten), Entgleisungen des Salzhaushaltes im Blut sowie Medikamente, die zu einer Verlängerung der QT-Zeit führen können (siehe Links).
Spezielle Behandlungsstrategien
Zu den speziellen Behandlungsstrategien zählt die Gabe von Betarezeptorenblockern. Allein durch die prophylaktische Behandlung mit Betarezeptorenblockern lässt sich bei etwa 75% der betroffenen Kinder im Vergleich zu nicht behandelten Betroffenen klinisch ein befriedigendes Ergebnis erzielen.
Die sogenannte genspezifische Therapie gehört zu den neueren speziellen Therapiemöglichkeiten für das Long QT-Syndrom.
Die Erweiterung der Therapiemaßnahmen in Form eines Stimulationsschrittmachers ist bei Patienten angezeigt, bei denen es ohne oder mit einem Betarezeptorenblocker zu einer definierten Erniedrigung der Herzfrequenz (Bradykardie) kommt und/ oder bradykardieabhängige Störungen auftreten. Besonderer Nutzen einer Schrittmacherstimulation ist bei LQT3-Patienten zu erwarten, da bei diesen die ausgeprägtesten QT-Verlängerungen im Rahmen von Bradykardien in Ruhe oder im Schlaf zu beobachten sind und diese durch eine einfache Frequenzanhebung am wirkungsvollsten zu beseitigen sind.
Für Patienten die unter Prophylaxe mit einem Betarezeptorenblocker wiederholt (symptomatische) Herzrhythmusstörungen zeigen oder für Patienten nach Reanimation stehen heute Interne Cardioverter Defibrillatoren (ICD) zur Verfügung. Diese ICD-Systeme enthalten zusätzlich eine Stimulationseinheit mit der eine Mindestfrequenz des Herzens sichergestellt werden kann.
Festzuhalten ist, dass sowohl Patienten mit einem Stimulationsschrittmacher als auch Patienten mit einem ICD selbstverständlich weiter einen Betarezeptorenblocker einnehmen müssen, um überhaupt dem Auftreten von Herzrhythmusstörungen vorzubeugen.
Eine weitere, heute jedoch insbesondere bei Kindern eher selten durchgeführte Maßnahme, stellt die linksseitige kardiothorakale sympathische Denervation (LCSD) dar. Ihr liegt die Vorstellung einer ungleichseitigen sympathischen Stimulation des Herzens beim Long QT-Syndrom zugrunde, die zu einer verstärkten Imbalanz der ventrikulären Repolarisation führt. Die LCSD soll eine Erhöhung der Schwelle für das Auftreten lebensbedrohlicher Herzrhythmusstörungen bewirken.