AG Lohmann

Volker Lohmann
Deputy Head of the Department
"MOLECULAR VIROLOGY"
Head of section "Virus-host interactions"
Replication of positive strand RNA viruses and their recognition by the cell intrinsic innate immune response
Phone: +49 (0)6221-56 6449
We are interested in different aspects of Hepatitis C virus (HCV) replication including the understanding of cell culture adaptation, structure-function analyses on the HCV polymerase and the identification of host cell factors engaged in HCV replication, e.g. the cellular lipid kinase PI4KA, Cyclophilins and microRNA 122. One of our focuses is a general understanding of the biogenesis of the membranous replication organelles of positive strand RNA viruses, in particular HCV and Norovirus. We are furthermore interested how HCV induces and counteracts innate and adaptive immune responses to finally achieve persistence, in contrast and comparison to hepatitis A virus infections, sharing a similar replication strategy, but always being cleared.

Research Team Members




Research Projects
I. Regulation of lipid homeostasis at the HCV replication organelle and its role in genome replication and pathogenesis
Hepatitis C virus (HCV) replication takes place at distinct vesicular membrane structures which are induced by a concerted action of the viral nonstructural proteins and host factors. In previous studies we have identified an intimate connection of HCV replication and the cellular lipid kinase Phosphatidylinositol-4-kinase IIIa, (PI4KIIIa, PI4KA). PI4KA converts phosphatidylinositol to phosphatidylinositol 4-phosphate (PI4P). The enzymatic activity of PI4KA is activated by the HCV nonstructural proteins NS5A and NS5B, resulting in elevated intracellular PI4P levels (Reiss et al., Cell Host & Microbe, 2011, Plos Path, 2013; Harak et al., JVI; 2014). This increased concentration of PI4P has a central role for the biogenesis of the viral replication organelle by shaping its lipid composition due to the action of lipid transport proteins like OSBP or FAPP2. However, activation of PI4KA in Huh7 hepatoma cells is deleterious for most HCV wildtype isolates due to an excess of PI4P, explaining the inefficient replication of patient derived viruses in cell culture. Surprisingly, we could demonstrate that replication enhancing adaptive mutations, which are essential to support HCV replication in hepatoma cells, in fact rely on a loss of function mechanism, abrogating activation of PI4KA to compensate for high PI4KA expression levels in hepatoma cells compared to primary hepatocytes (Harak et al., Nature Microbiology, 2016). Based on this finding we could establish a regimen based on PI4KA/Casein Kinase Iα (CKIα) inhibitors, allowing efficient replication of HCV wt isolates in cell culture. Currently we are aiming at a comprehensive understanding of the mechanisms involved in shaping the lipid composition at the HCV replication organelle. We further investigate the mechanism of action of Sec14L2, a lipid transporter protein expressed in hepatocytes but not in Huh7. Sec14L2 was recently identified by others to stimulate replication of HCV wt isolates, but the mechanism is still poorly defined and the effect is very variable for different clinical isolates (Costa et al., J. Hepatol., 2018). We are currently testing the hypothesis that the lipid transport function of Sec14L2 might link its mechanism of action to PI4KA.
Another independent project is dedicated to the impact of elevated PI4P levels, which appear as a consequence of PI4KA activation by HCV, to viral pathogenesis. PI4P is a precursor of other phosphoinositides involved in various cellular signaling processes, and by itself has crucial functions in intracellular vesicular transport. In addition, PI4KA expression is upregulated in hepatoma cells and therefore might contribute to cancer growth. We therefore use different cell based and animal models to understand the impact of PI4KA abundance and activation on cellular signaling cascades, cell growth and morphology to unravel potential mechanistic links to hepatocellular carcinoma.
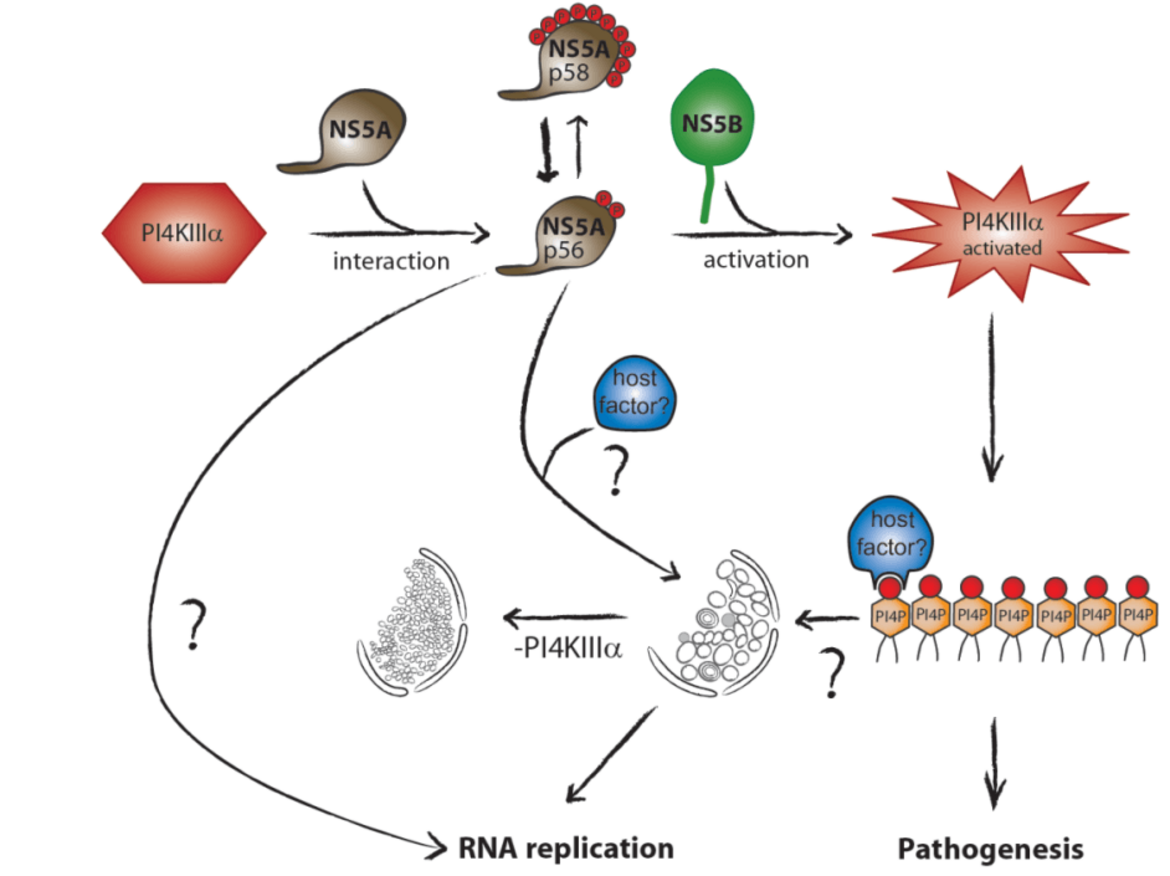
Activation of the cellular lipid kinase PI4KIIIa (PI4KA) by the HCV nonstructural proteins NS5A and NS5B results in an intracellular accumulation of PI4P, which is crucial for shaping the lipid composition of the viral replication organelle and affects several cellular processes potentially contributing to viral pathogenesis.
II. The role of miR-122 in HCV translation and replication
The liver specific microRNA 122 (miR-122) recognizes two conserved sites at the 5’ end of the hepatitis C virus (HCV) genome and contributes to stability, translation and replication of the viral RNA. Stimulation of the HCV internal ribosome entry site (IRES) by miR-122 is essential for efficient viral replication. We have shown that the mechanism relies on a dual function of the 5’ terminal primary sequence in the complementary positive (IRES-mediated translation) and negative strand (RNA replication), requiring different secondary structures. Our results suggest that miR-122 binding assists the folding of a functional IRES in an RNA chaperone-like manner by suppressing energetically favorable alternative secondary structures (Schult et al., Nature Comm., 2018). We are currently assessing the quantitative contribution of the various functions of miR-122 in HCV translation, genome stability and replication in a mathematical model of HCV replication (Binder et al., Plos Path, 2013). We further aim to obtain a comprehensive understanding of the spatio-temporal regulation of the interaction between miR-122 and the viral genome.
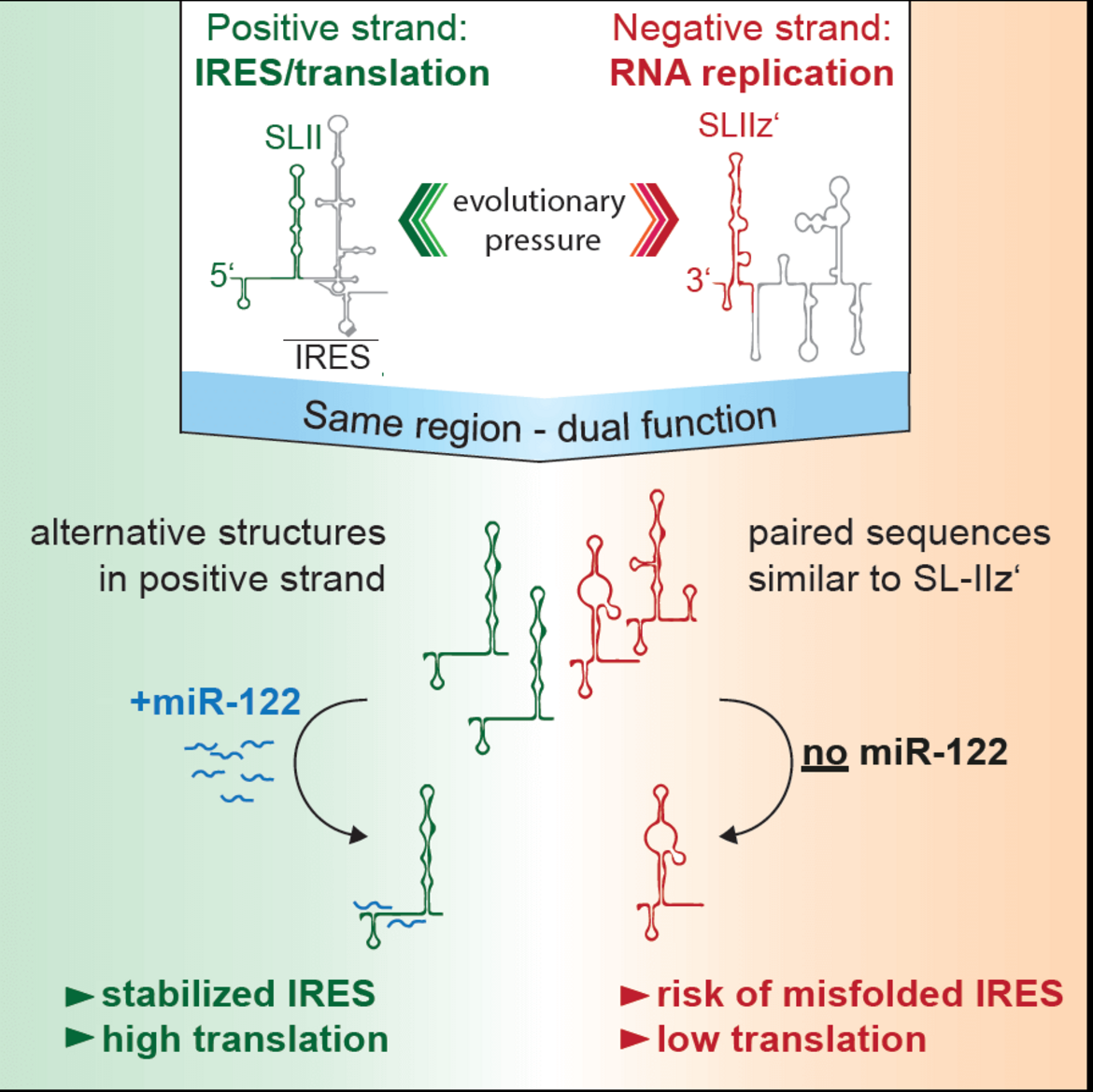
Schematic of the mechanism how mir-122 contributes to the stimulation of HCV translation. Binding of mir-122 to the 5’UTR of the HCV genome stabilizes the folding of the internal ribosome entry site (IRES) by preventing alternative structures, which are energetically favorable due to the dual function of the same sequence in translation (positive strand) and replication (negative strand), requiring different RNA secondary structures (Schult et al., Nat. Comm., 2018).
III. Understanding the Toll-like receptor 3 response against HCV and HAV and its contribution to persistence and clearance of viral infections
Hepatitis C virus (HCV, family Flaviviridae) and hepatitis A virus (HAV, family Picornaviridae) are both positive strand RNA viruses with very similar mechanisms of RNA replication. However, HAV infections are always cleared whereas HCV establishes persistence in ca. 70% of infected individuals. Clearance correlates with low levels of innate immune responses in the liver in case of HAV, whereas persistence in case of HCV is associated with a long-lasting induction of a variety of interferon-induced genes (ISGs), which mainly originate from infected cells. Still, both viruses activate a similar panel of pattern recognition receptors and have evolved identical countermeasures involving cleavage of the same adaptor molecules crucial for mounting intracellular innate immune responses. Our previous work has established replication models for both viruses allowing a thorough side-by-side comparison (Esser-Nobis et al., Hepatology 2015). HAV, in stark contrast to HCV, does not induce Toll-like receptor 3 (TLR3)-mediated innate immune responses, despite the fact that the signaling pathway is not efficiently blocked in cells infected with HAV. This result points to substantial differences in the constitution of the viral replication compartment and the fate of viral replication intermediates, reaching TLR3 in case of HCV, but not in case of HAV. However, HCV has established an escape mechanism to dampen TLR3 responses by secretion of replication intermediates in extracellular vesicles, suggesting that a balanced TLR3 response might support the establishment of a persistent infection (Grünvogel et al., Gastroenterology, 2018). We are currently aiming at a molecular understanding of the fate of the double stranded replication intermediates of both viruses. In addition, we are interested in general aspects of the TLR3 response in hepatocytes. To this end, we are analyzing the function of various TLR3 polymorphisms found in the human population. We are furthermore analyzing hits from a Cripr/Cas9 based knockout screen on the TLR3 pathway to identify novel factors involved in its regulation.
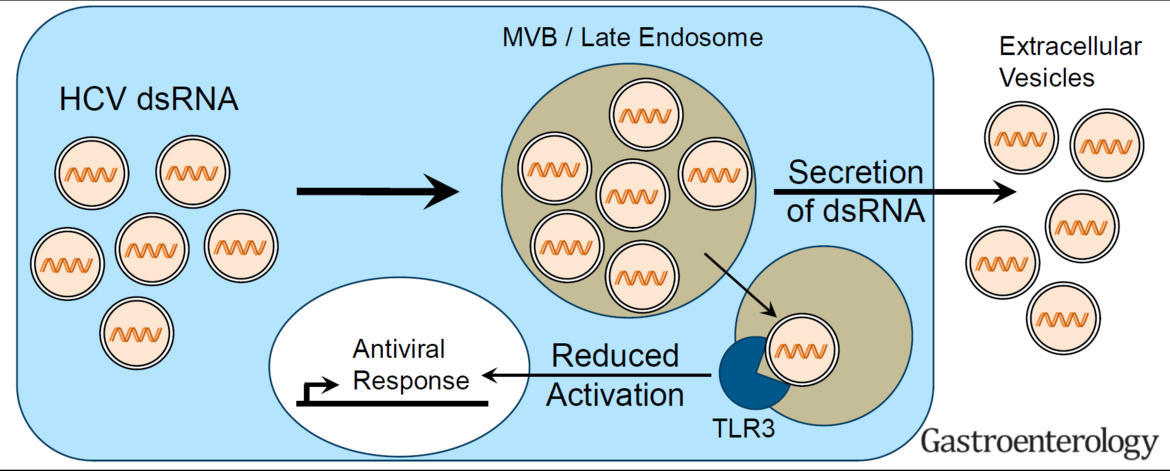
HCV partially escapes recognition by TLR3 upon secretion of double stranded replication intermediates in exosomes (Grünvogel et al., Gastroenterology, 2018).
IV. Function of norovirus nonstructural proteins
Human noroviruses (huNoV) are the most frequent cause of non-bacterial acute gastroenteritis worldwide, particularly genogroup II genotype 4 (GII.4) variants. The viral nonstructural (NS) proteins encoded by the ORF1 polyprotein induce vesical clusters harboring the viral replication sites. In a previous study we compared the ultrastructural changes induced by expression of norovirus ORF1 polyproteins with those induced upon infection with murine norovirus (MNV). Characteristic membrane alterations induced by ORF1 expression resembled those found in MNV infected cells, consisting of vesicle accumulations likely built from the endoplasmic reticulum (ER) which included single membrane vesicles (SMVs), double membrane vesicles (DMVs) and multi membrane vesicles (MMVs). Expression of GII.4 NS1-2, NS3 and NS4 fused to GFP each revealed distinct membrane alterations. Interestingly, NS4 was the only GII.4 protein capable of inducing SMV and DMV formation when expressed individually. We thereby identified NS4 as a key factor in the formation of membrane alterations of huNoV and provided models of the putative membrane topologies of NS1-2, NS3 and NS4 to guide future studies. Currently we are analyzing the specific functions of these proteins in the biogenesis of the human norovirus replication organelle. We are further aiming at the establishment of robust cell culture models for human noroviruses.
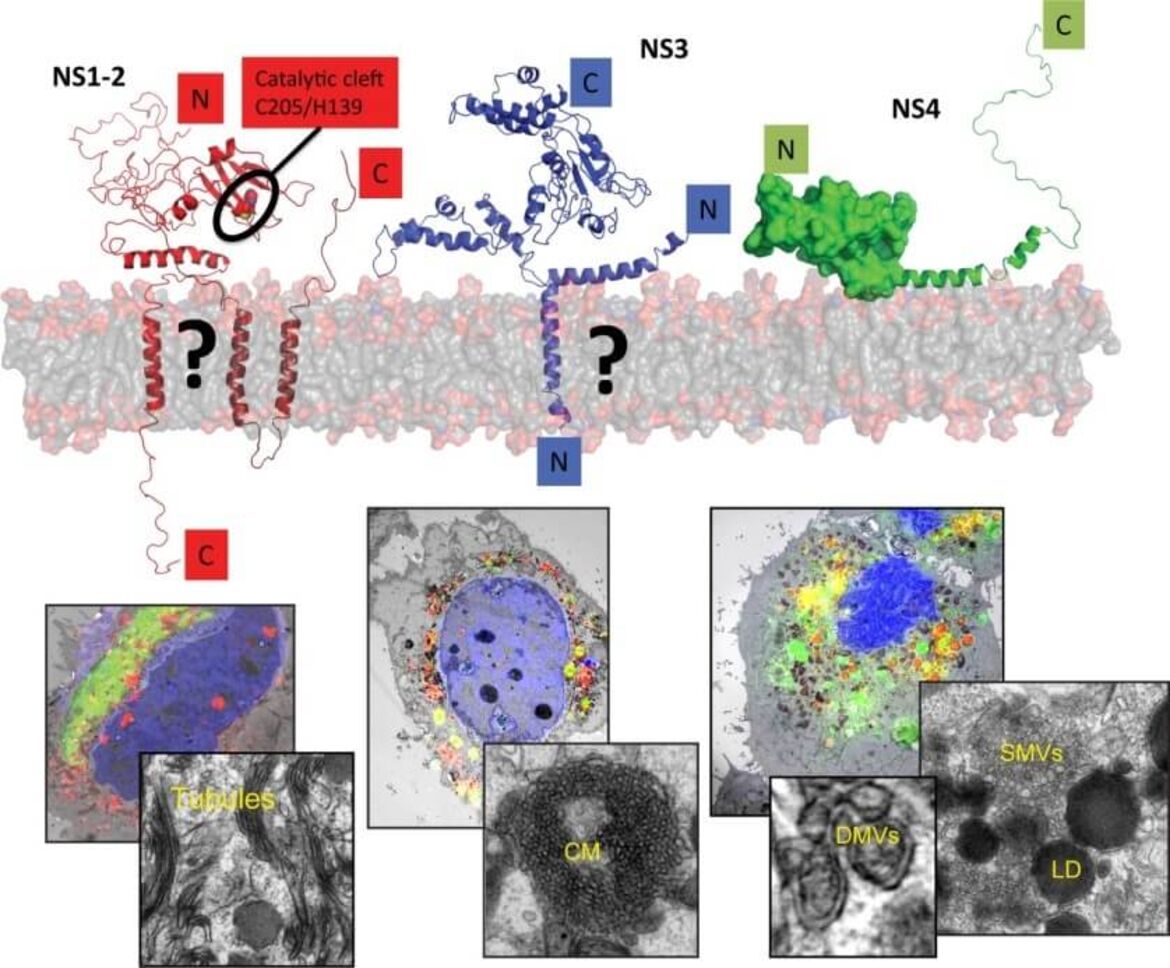
Replication of noroviruses is linked to membrane alterations induced by the nonstructural proteins. The main drivers of this process are NS1-2, NS3 and NS4. The upper panel provides a tentative view on the proposed structure of these proteins, the lower panels a correlative-light and electron microscopy study to identify distinct membrane alterations induced by the individually expressed proteins (Doerflinger et al., Plos Path, 2017).
Funding




V. Major publications
Tran CS, Kersten J, Yan J, Breinig M, Huth T, Poth T, Colasanti O, Riedl T, Faure-Dupuy S, Diehl S, Verhoye L, Li TF, Lingemann M, Schult P, Ahlén G, Frelin L, Kühnel F, Vondran FWR, Breuhahn K, Meuleman P, Heikenwälder M, Schirmacher P, Bartenschlager R, Laketa V, Roessler S, Tschaharganeh DF, Sällberg M, Lohmann V.: Phosphatidylinositol 4-kinase III alpha governs cytoskeletal organization for invasiveness of liver cancer cells. Gastroenterology. 2024 Apr 16:S0016-5085(24)00424-4. doi: 10.1053/j.gastro.2024.04.009. Epub ahead of print. PMID: 38636680.
Colasanti O, Burm R, Huang HE, Riedl T, Traut J, Gillich N, Li TF, Corneillie L, Faure-Dupuy S, Grünvogel O, Heide D, Lee JY, Tran CS, Merle U, Chironna M, Vondran FFW, Esser-Nobis K, Binder M, Bartenschlager R, Heikenwälder M, Meuleman P, Lohmann V. Comparison of HAV and HCV infections in vivo and in vitro reveals distinct patterns of innate immune evasion and activation. J Hepatol. 2023 Sep;79(3):645-656. doi: 10.1016/j.jhep.2023.04.023. Epub 2023 Apr 29. Erratum in: J Hepatol. 2024 Jan;80(1):171-172. PMID: 37121436.
Chen P, Wojdyla JA, Colasanti O, Li Z, Qin B, Wang M, Lohmann V, Cui S. Biochemical and structural characterization of hepatitis A virus 2C reveals an unusual ribonuclease activity on single-stranded RNA. Nucleic Acids Res. 2022 Sep 9;50(16):9470-9489. doi: 10.1093/nar/gkac671. PMID: 35947700; PMCID:PMC9458454.
Heuss C, Rothhaar P, Burm R, Lee JY, Ralfs P, Haselmann U, Ströh LJ, Colasanti O, Tran CS, Schäfer N, Schnitzler P, Merle U, Bartenschlager R, Patel AH, Graw F, Krey T, Laketa V, Meuleman P, Lohmann V. A Hepatitis C virus genotype 1b post-transplant isolate with high replication efficiency in cell culture and its adaptation to infectious virus production in vitro and in vivo. PLoS Pathog. 2022 Jun 28;18(6):e1010472. doi: 10.1371/journal.ppat.1010472. PMID: 35763545; PMCID: PMC9273080.
Grünvogel O, Colasanti O, Lee JY, Klöss V, Belouzard S, Reustle A, Esser‐Nobis K, Hesebeck‐Brinckmann J, Mutz P, Hoffmann K, Mehrabi A, Koschny R, Vondran FWR, Gotthardt D, Schnitzler P, Neumann‐Haefelin C, Thimme R, Binder M, Bartenschlager R, Dubuisson J,Dalpke AH, Lohmann V: Secretion of Hepatitis C Virus Replication Intermediates Reduces Activation of Toll‐Like Receptor 3 in Hepatocytes. Gastroenterology 2018; 154(8):2237‐2251
Schult P, Roth H, Adams RL, Mas C, Imbert L, Orlik C,Ruggieri A, Pyle AM, Lohmann V: microRNA‐122 amplifies hepatitis C virus translation by shaping the structure of the internal ribosomal entry site. Nat Commun 2018; 4; 9(1):2613
Doerflinger SY, Cortese M, Romero‐Brey I, Menne Z, Tubiana T, Schenk C, White PA, Bartenschlager R, Bressanelli S, Hansman GS, Lohmann V: Membrane alterations induced by nonstructural proteins of human norovirus. PLoS Pathog 2017; 27;13(10)
Harak C, Meyrath M, Romero‐Brey I, Schenk C, Gondeau C, Schult P, Esser‐Nobis K, Saeed M, Neddermann P, Schnitzler P, Gotthardt D, Perez Del‐Pulgar S, Neumann‐Haefelin C, Thimme R, Meuleman P, Vondran FW, Francesco R, Rice CM, Bartenschlager R, Lohmann V:Tuning a cellular lipid kinase activity adapts hepatitis C virus to replication in cell culture. Nat Microbiol. 2016 Dec 19;2:16247.
Lohmann V., Körner F, Koch JO, Herian U, Theilmann L and Bartenschlager R.: Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999; 285: 110‐113