AG Seitz

Stefan Seitz
"MOLECULAR VIROLOGY"
Phone: +49 (0)6221-56 4225
Fax: +49 (0)6221-56 4570
Research Team Members




Research Projects
I. DYNAMICS OF HEPATITIS B VIRUS MORPHOGENESIS AND ITS ALTERATIONS DURING CHRONIC INFECTION
Chronic hepatitis B is the major driver for the development of liver cirrhosis and primary liver cancer globally accounting for more than 800 thousand deaths per year. In the serum of patients chronically infected with the hepatitis B virus (HBV) one does not only find complete DNA-containing virions, but also virions harboring “empty” or RNA-containing capsids. The proportion of these non-canonical forms of virions in chronic carriers varies drastically depending on the intrahepatic viral replication activity and the treatment status. Currently, there is little known about the ultrastructural and biochemical foundations of particle morphogenesis giving rise to these non-canonical particles.
In earlier research work we identified two distinct morphotypes of hepatitis B virions in the blood circulation (Seitz et al., 2007). Based on this observation, we subsequently discovered a unique maturation step that slowly renders secreted virions infectious: Virions become released from the host cell in an inactive form and successively develop infectivity due to the translocation of their major receptor binding determinant across the viral membrane onto the outer particle surface (Seitz et al., 2016).
These findings led us to propose that HBV particles are subject to defined ultrastructural dynamics with a series of thermodynamically driven, temporally and spatially regulated events during particle morphogenesis (Fig. 1, reviewed in Seitz et al., 2020). This might be key to determining the fate of nucleocapsids in the host cell by deciding between either secretion as enveloped virions or recycling to the nucleus to sustain the pool of viral covalently closed circular DNA, which is the central reservoir for lifelong HBV persistence.
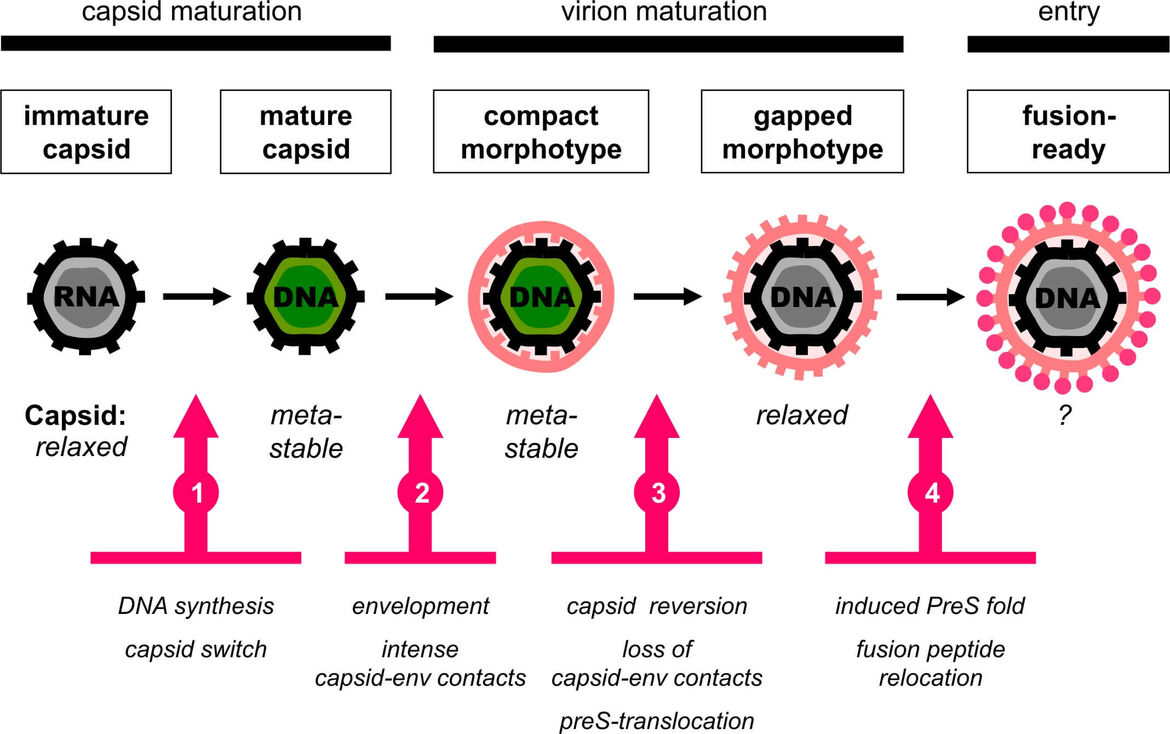
In order to dissect the proposed chain of events, we will characterize nucleocapsids containing the authentic replication complex by biochemical and structural biology methods. The nucleocapsids will be arrested in different maturation states and subjected to structure determination by cryo-electron microscopy. We will investigate the impact of any structural changes on the binding competence of nucleocapsids to surface protein segments as prerequisite for capsid envelopment. Moreover, we perform thorough biochemical interaction studies to quantitatively describe the binding kinetics of nucleocapsids to the HBV surface glycoproteins. Nuclear magnetic resonance techniques will be applied to resolve the structure of surface protein segments bound to nucleocapsids. The insight gained into structural details and the biochemical parameters of the interactions determining nucleocapsid envelopment will allow us the quantitative characterization and modelling of the pathways of HBV particle morphogenesis with respect to their variability during chronic infection.
Collaboration partners:
- Anne Schütz, Department of Chemistry, LMU Munich
- Simone Mattei, EMBL Heidelberg
- Walter Mier, Nuclear Medicine, University Clinics, Heidelberg
- Ursula Klingmüller, DKFZ, Systems Biology of Signal Transduction (B200)
References:
- Harati Taji Z., Bielytskyi P., Shein M., Sani M.A., Seitz S.*, and Schütz A.K.* (2022). Transient RNA Interactions Leave a Covalent Imprint on a Viral Capsid Protein. J. Am. Chem. Soc. 144, 8536-8550. PMID: 35512333
*Co-corresponding authors - Seitz S., Habjanič J., Schütz A.K., and Bartenschlager R. (2020). The Hepatitis B Virus Envelope Proteins: Molecular Gymnastics Throughout the Viral Life Cycle. Annu. Rev. Virol. 7, 263-288. PMID: 32600157
- Seitz S., Iancu C., Volz T., Mier W., Dandri M., Urban S., and Bartenschlager R. (2016). A Slow Maturation Process Renders Hepatitis B Virus Infectious. Cell Host Microbe. 20, 25-35. PMID: 27321908
- Seitz S., Urban S., Antoni C., and Böttcher B. (2007). Cryo-electron microscopy of hepatitis B virions reveals variability in envelope capsid interactions. EMBO J. 26, 4160-7. PMID: 17762862
II. INCREASING PANDEMIC PREPAREDNESS BY COMPUTATIONAL HIGH-THROUGHPUT VIRUS DISCOVERY: IDENTIFICATION OF RNA VIRUSES AND HOST RESERVOIRS WITH HIGH SPILLOVER RISK
We have a long-standing interest in the discovery and evolution of viruses in general. Our “Data-Driven Virus Discovery” (DDVD; Lauber & Seitz, 2022) strategy involves two computational pipelines, termed Virushunter and Virusgatherer, dedicated to the identification of Next-Generation sequencing (NGS) experiments positive for viral sequence reads and the subsequent assembly of the corresponding viral genomes, respectively (Fig. 2).
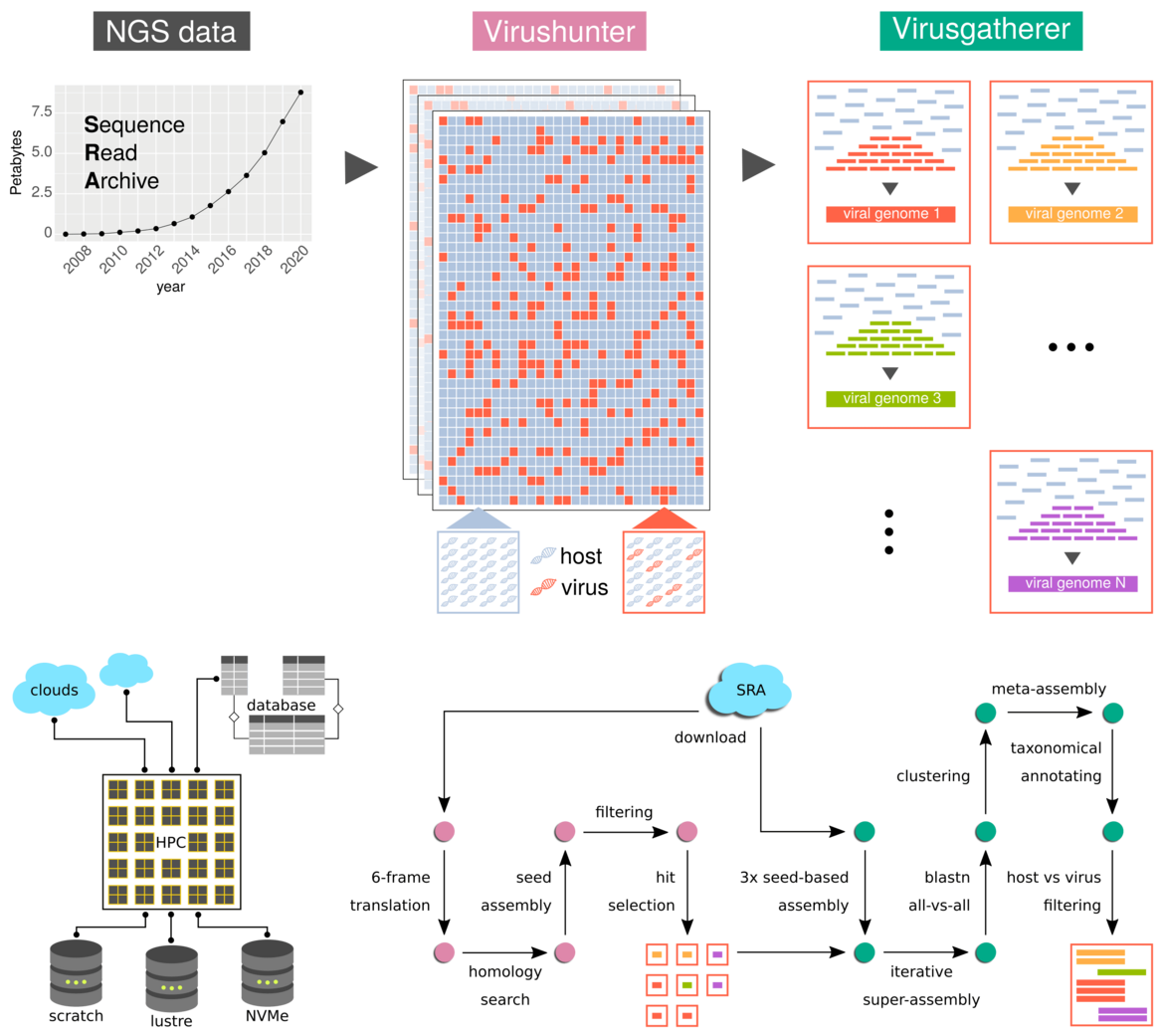
In this project we have screened >500,000 individual transcriptome sequencing datasets covering the whole spectrum of eukaryotic organisms for the presence of sequences of viral origin. We took advantage of the fact that the genomes and transcripts of known and unknown viruses are sequenced incidentally as “bycatch”, if the source material stems from an organ, tissue or cell infected by a virus at the time of sampling. In total, we recovered ~150,000 contigs representing genomic sequences of bona fide eukaryotic RNA viruses. About two thirds of them are only distantly related to any known RNA viruses constituting whole new virus families (Fig. 3).
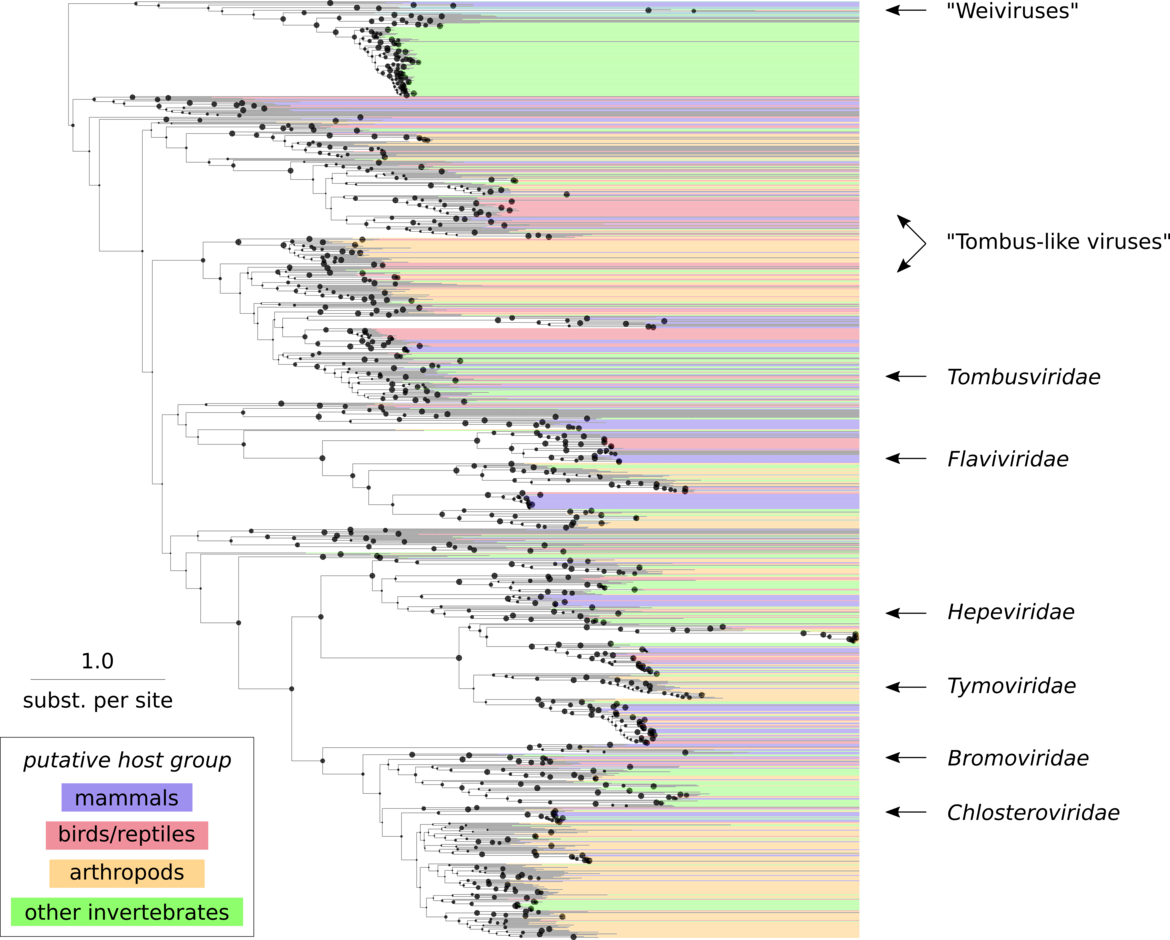
We will now use these data in order to contribute to sustainable strategies towards preparedness for future emerging infectious diseases and pandemics. We will derive, validate and apply a spillover and pandemic risk score to identify RNA viruses with highest zoonosis probability. We expect that this work will provide the basis for creating platforms for the high-throughput development of diagnostics, vaccines and antivirals for high-risk candidate virus groups.
Collaboration partners:
- Chris Lauber, Institute for Experimental Virology, TWINCORE Centre for Experimental and Clinical Infection Research, Hannover.
- Pascal Mutz, NIH, Bethesda MD, USA
References:
- Lauber C., and Seitz S. (2024). Deep mining of the Sequence Read Archive reveals major genetic innovations in coronaviruses and other nidoviruses of aquatic vertebrates. PLoS Pathog. 20(4):e1012163.doi: 10.1371. PMID: 38648214
- Lauber C., and Seitz S. (2022). Opportunities and Challenges of Data-Driven Virus Discovery. Biomolecules 12, 1073. PMID: 36008967
- Lauber C., Seifert M., Bartenschlager R., and Seitz S. (2019). Discovery of highly divergent lineages of plant-associated astro-like viruses sheds light on the emergence of potyviruses. Virus Res. 260, 38-48. PMID: 30452944
- Lauber C.*, Seitz S.*, **, Mattei S., Suh A., Beck J., Herstein J., Börold J., Salzburger W., Kaderali L., Briggs J.A.G., and Bartenschlager R. (2017). Deciphering the Origin and Evolution of Hepatitis B Viruses by Means of a Family of Non-enveloped Fish Viruses. Cell Host Microbe. 22, 387-399.e6. PMID: 28867387
*Equal contribution, **Corresponding author
III. TRACING THE ORIGIN AND EVOLUTION OF HEPATITIS B-RELATED VIRUSES – A MODEL SYSTEM TO COMPUTATIONALLY DERIVE VIRAL SPILLOVER RISK SCORES
The pilot study in which we developed and applied our above described bipartite Virushunter-Virusgatherer computational pipeline was dedicated to the discovery of viruses related to the human hepatitis B virus (HBV, family Hepadnaviridae). By screening >25,000 NGS datasets available in NCBI’s Sequence Read Archive (SRA) at that time, we were able to identify 21 novel viral species in vertebrates, 17 of them the very first hepatitis B-like viruses found in bony fish. Surprisingly, 13 of these fish viruses lacked the typical envelope protein gene and thus constituted a novel sister family of hepadnaviruses which we termed nackednaviruses. With this collection of viral genomes we were able to reliable root the hepadnaviral phylogeny. A time calibration based on endogenous hepadnaviral elements integrated into the genomes of song birds revealed that both sister families diverged from a common ancestor in the Silurian ~430 million years ago (mya). The hepadnaviral lineage had acquired its characteristic envelope protein gene ~370 mya at about the same time when the first terrestrial vertebrates (tetrapods) evolved.
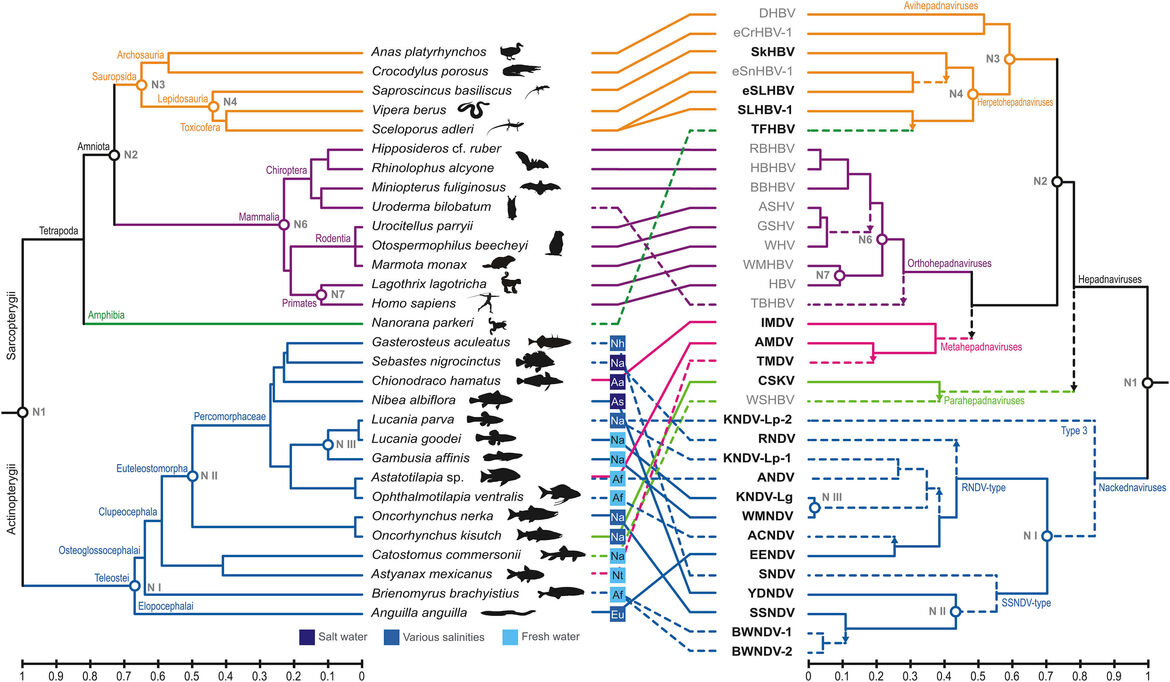
Remarkably, we observed quite distinct patterns of virus-host association: While the viruses infecting tetrapods underwent a long-term co-evolution in intimate association with their respective hosts, the fish viruses frequently jump between hosts (Fig. 4). Through regular search updates, our virus taxon sampling meanwhile further grew to almost 100 novel viral genomes. In many cases we find closely related nackednaviruses infecting distantly related fish species, further corroborating their inherent tendency to repeated host-switches. Thus, the nackedna-/hepadnavirus sister groups represent an optimal model system to develop and test computational approaches for deriving viral spillover risk scores that shall subsequently be applied to the tens of thousands novel RNA viruses in order to contribute to future pandemic preparedness (see above).
Collaboration partners:
- Chris Lauber, Institute for Experimental Virology, TWINCORE Centre for Experimental and Clinical Infection Research, Hannover.
- Pascal Mutz, NIH, Bethesda MD, USA
- Simone Mattei, EMBL Heidelberg
References:
- Lauber C.*, Seitz S.*, **, Mattei S., Suh A., Beck J., Herstein J., Börold J., Salzburger W., Kaderali L., Briggs J.A.G., and Bartenschlager R. (2017). Deciphering the Origin and Evolution of Hepatitis B Viruses by Means of a Family of Non-enveloped Fish Viruses. Cell Host Microbe. 22, 387-399.e6. PMID: 28867387
*Equal contribution, **Corresponding author - Beck J., Seitz S., Lauber C., and Nassal M. (2021). Conservation of the HBV RNA element epsilon in nackednaviruses reveals ancient origin of protein-primed reverse transcription. Proc. Natl. Acad. Sci. U S A. 118, e2022373118. PMID: 33753499
- Glebe D.*, Goldmann N., Lauber C., and Seitz S.* (2021). HBV evolution and genetic variability: Impact on prevention, treatment and development of antivirals. Antiviral Res. 186, 104973. PMID: 33166575
*Co-corresponding authors
IV. ELUCIDATING THE ROLE OF ANELLOVIRUSES IN HUMAN PATHOGENESIS
Anelloviruses are a highly diverse group of non-enveloped viruses with circular single-stranded DNA genomes ranging between 2.0-3.8 kb. These viruses are ubiquitously present in humans and other vertebrates where they establish persistent infections. Currently, their potential role in development of human diseases, particularly cancer and autoimmune disorders, remains elusive.
We utilize our high-throughput computational pipeline for virus discovery in order to detect anellovirus sequences both in publicly accessible NGS data and in protected NGS data from cancer patients to which we get exclusive access through collaboration partners in Heidelberg.
Frequently, we detect several family members simultaneously within the same sequencing sample. From these observations, we conclude that anelloviruses build viral communities or swarms composed of distinct viral lineages co-occurring in one and the same host and tissue (Fig. 5). Thus, it is possible that it is a dysbalance in the composition of such communities rather than the mere presence of these viruses that contributes to pathogenesis. We hypothesize that, although in many cases not causing harm over years or decades, emerging dysbalances in the structure of these complex viral swarms might drive disease onset and progression.
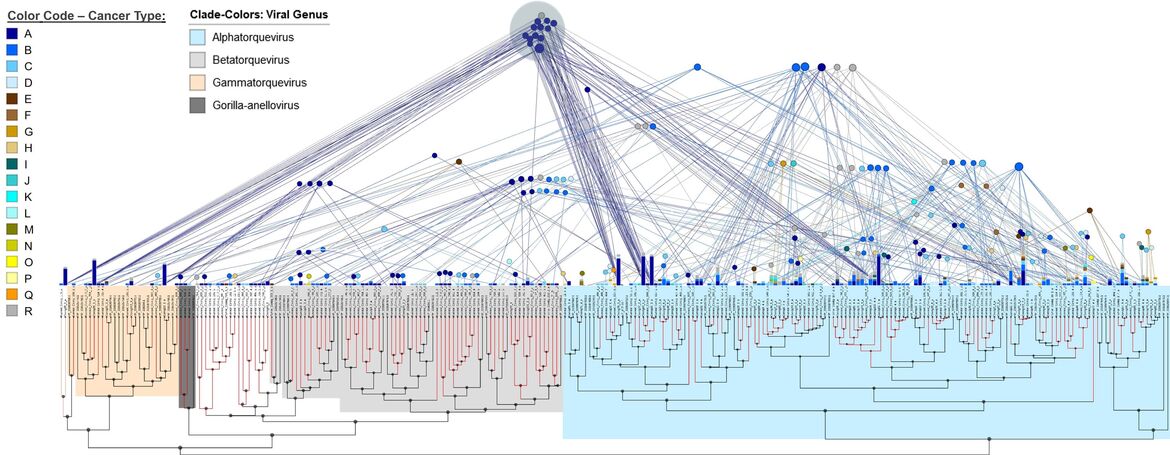
To this end, it is not sufficient just to identify novel viral sequences but their occurrence has to be thoroughly correlated to i) the disease conditions between infected individuals, ii) the course of disease (progression) within an individual and iii) between healthy and pathological tissues. Currently, we aim at a comprehensive description of the spectrum of human anelloviruses by our screening approach. We will then perform computational and laboratory analyses to reveal i) the disease association of identified viral species and subgroups, ii) the presence of viral oncogenes and iii) epidemiological data regarding the viral prevalence and the related risk of disease.
The controlled NGS data provided by our internal collaboration partners create an ideal opportunity to tackle these questions. This project is a proof-of-concept study that can be extended to any kind of DNA and RNA viruses in order to unmask so far hidden oncoviruses.
Our collaboration partners are:
- Peter Lichter & Marc Zapatka, Molecular Genetics, DKFZ, Heidelberg
- Stefan Pfister & Natalie Jäger, Pediatric Neurooncology, DKFZ, Heidelberg