



Arbeitsgruppe Prof. Dr. Hadaschik


Research Focus
Our group focusses on the immunology of autoimmune skin diseases.
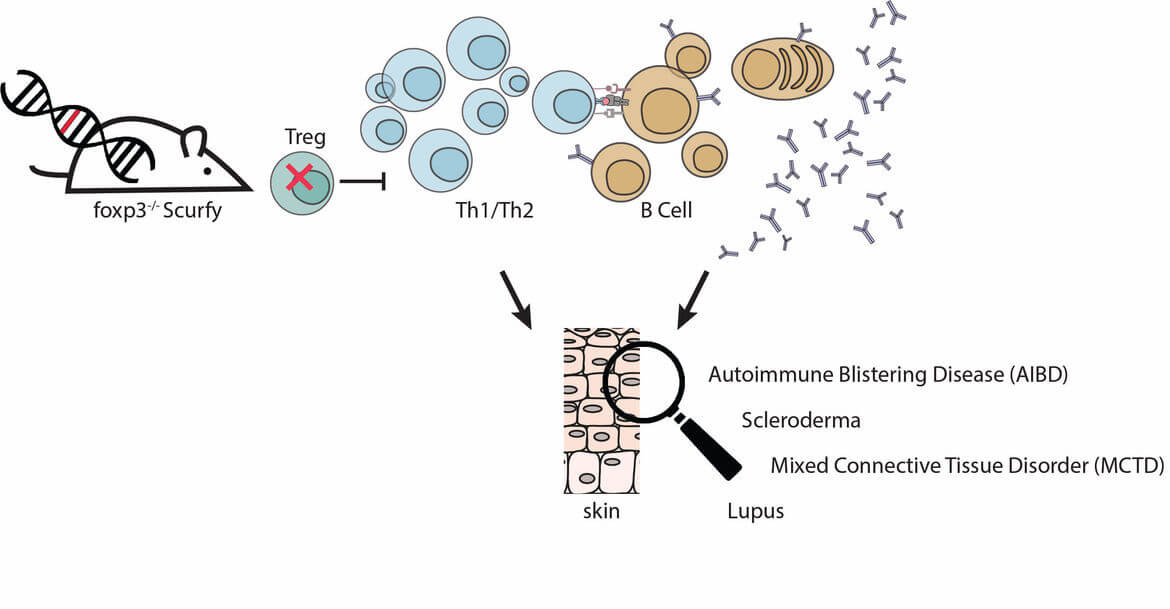
In a healthy organism the activity of autoreactive CD4+ T cells is usually suppressed by regulatory T cells (Treg) to protect against autoimmunity. Tregs are generated in the thymus, circulate in the periphery where they suppress activated autoreactive CD4+ T cells, which have escaped thymic deletion, to maintain peripheral tolerance.
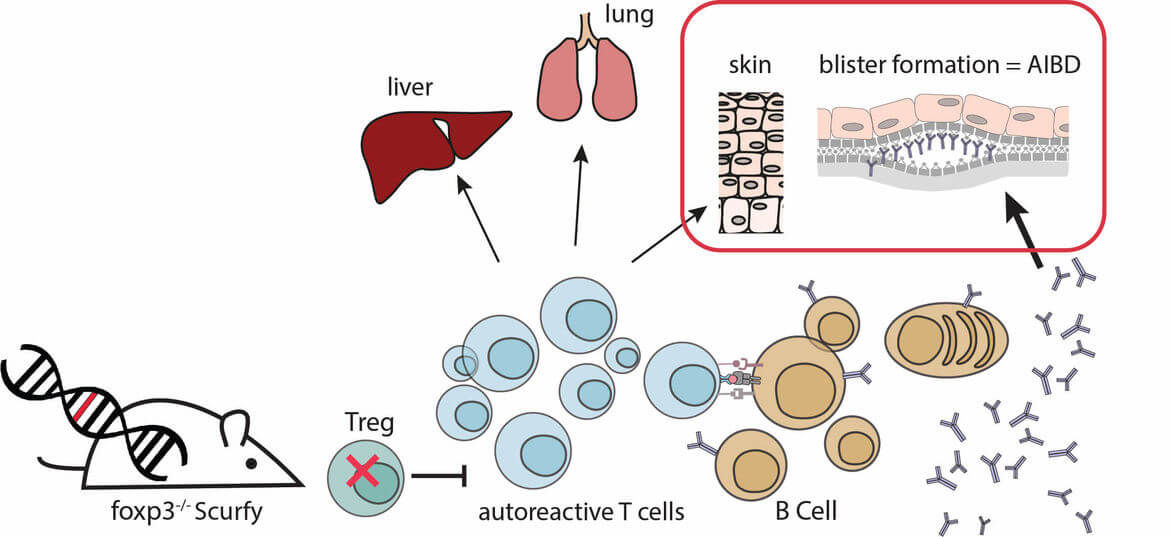
Scurfy mice have a mutation in the FoxP3 gene, which is crucial for the function of regulatory T cells. The missing Treg-control leads to uncontrolled proliferation of autoreactive CD4+ T cells. Scurfy mice spontaneously develop CD4+ T cell-mediated autoimmune inflammation finally leading to multiorgan failure. Interestingly, all Scurfy mice develop severe erosive skin disease. In addition, patients with the rare “immunodysregulation, polyendocrinopathy, enteropathy, x-linked (IPEX) syndrome”, caused by the same functional defect in Treg as in Scurfy mice, suffer from similar symptoms as scurfy mice: They develop multiorgan autoimmune inflammation and the majority (>70%) of patients develops severe skin disease.
The autoimmune disease in Scurfy mice occurs spontaneously, so that it reproduces the pathogenesis of autoimmune reactions in the human system. Therefore, the Scurfy mouse is a suitable model to investigate the control function of Treg for autoimmune-mediated skin diseases in humans.
We are interested in the autoimmune disease, which develops in the absence of functional of regulatory T cells.
Projects
Autoimmune Blistering Disease in the Absence of Regulatory T cells
Autoimmune blistering diseases (AIBD), such as bullous pemphigoid (BP) or pemphigus vulgaris (PV), are severe autoimmune diseases of the skin that are caused by autoantibodies against structural proteins of the skin. The binding of autoantibodies in the skin leads to blistering and depending on the location of blister formation the different AIBDs are diagnosed. This project aims at analyzing the role of regulatory T cells for the pathophysiology of AIBD.
We previously could demonstrate that AIBDs spontaneously develop in the absence of functional regulatory T cells using the scurfy mouse model. Scurfy mice lack Tregs due to a missense mutation in their transcription factor foxp3 and suffer from severe skin inflammation with a Th1/Th2 differentiation of the immune infiltrate. In addition scurfy mice show high titers of autoantibodies against skin proteins. In scurfy sera, we could detect autoantibodies against different autoantigens associated with several types of autoimmune blistering diseases (AIBD), such as BP180, Laminin 332, and Collagen VII. Histological examination of scurfy skin revealed the presence of subepidermal blisters. These blisters, together with the deposition of autoantibodies in the skin, demonstrate that scurfy mice develop the phenotype of AIBD (Haeberle et al 2018)
Currently, we are focusing on elucidating the interaction between autoreactive T and B cells and the resulting induction of autoantibody production.
Autoantigens in autoimmune skin disease in absence of Treg
We are interested in identifying potential new Autoimmune Blistering Disease (AIBD) - related autoantigens.
We successfully described Keratin 14 (K14) as a new autoantigen in the skin which is attacked by autoreactive T cells and B cells in the Scurfy mouse (Huter et al 2010). Patients with IPEX syndrome, harboring the same genetic defect as scurfy mice, also show reactivity to K14.
Additional, we isolated and characterized a spontaneously derived autoantibody from the scurfy mouse. This autoantibody is pathogenic in vivo and binds to BP230, a hemidesmosomal plaque protein and therefore displays clinical relevance (Haeberle et al 2018). Currently, we aim to clarify the pathomechanism of blister formation of BP230-specific autoantibodies and translate the findings to the clinic.
Role of Th2-associated cytokine IL-31 in Bullous Pemphigoid
Bullous Pemphigoid (BP) is the most common type of Autoimmune Blistering Disease and is characterized by subepidermal blister formation and erythema. Additional, patients suffer frequently of pruritus. The proinflammotry cytokine IL-31 is known to induce itch-signals and we could identify increased levels of IL-31 in sera and lesional skin of BP patients (Salz et al 2017).
We are investigating the effect of IL-31 blockade as a potential therapeutic approach in BP patients.
Differentiation Factors for Th2 Deviated Autoimmune skin disease in scurfy mice
The autoreactive CD4+ T cells in inflamed Scurfy skin show a strong Th1/Th2-polarization as indicated by their cytokine profile (IL4, IL5 and IL13) (Haeberle & Raker et al 2017). In this project we focus on the mechanisms of Th2 differentiation of CD4 T cells of scurfy mice. We identified thymic stromal lymphopoietin (TSLP) as potential differentiation factor. In addition the Th2-associated cytokine IL31 is differentially expressed in Scurfy skin in comparison to WT skin. The results of this project will broaden the understanding of the pathology of Th2-biased allergic disease as atopic dermatitis and asthma and might further impact on future therapeutic strategies.
Identification of anti-nuclear autoantibodies (ANAs) in scurfy mice
Lack of regulatory T cells in scurfy mice results in the expansion of autoreactive Th2 cells and autoantibody production. These autoantibodies are polyclonal, reactive for skin and other organs. However, we studied the presence of anti-nuclear autoantibodies in scurfy mice which are characteristic for different skin manifestations, e.g. lupus (Hadaschik et al 2015), scleroderma (Haeberle & Raker, 2017), mixed connective tissue disease (Yilmaz et al 2019).
Group members
Arbeitsgruppenleiterin
Postdocs
-

Dr. med. Torben Ramcke

Technische Assistentin
-

Lena Beideck
Former Lab Members
Ehemalige Kollegen
-

Dr. med. Mareen Zielonka







Selected publications
2020
Cheng X*, Haeberle S*, Shytaj IL , Gama-Brambila RA, Theobald J, Ghafoory S, Wölker J, Basu U, Schmidt C, Timm A , Taškova K, Bauer AS, Hoheisel J, Tsopoulidis N, Fackler OT, Savarino A, Andrade-Navarro MA, Ott I, Lusic M, Hadaschik EN, Wölfl S, NHC-gold compounds mediate immune suppression through induction of AHR-TGFβ1 signalling in vitro and in scurfy mice, Commun Biol, 2020 Jan 3;3:10. doi: 10.1038/s42003-019-0716-8. eCollection 2020.
2019
Yilmaz OK, Haeberle S, Zhang M, Fritzler MJ, Enk AH and Hadaschik EN. Scurfy Mice Develop Features of Connective Tissue Disease Overlap Syndrome and Mixed Connective Tissue Disease in the Absence of Regulatory T Cells. Front Immunol, 2019, 10, p881.
2018
Haeberle S, Wei X, Bieber K, Goletz S, Ludwig R, Schmidt E, Enk AH, Hadaschik EN. Treg deficiency leads to pathogenic BP230 autoantibody and autoimmune bullous disease, J Allergy Clin Immunol. 2018. pii: S0091-6749(18)30614-6. doi: 10.1016/j.jaci.2018.04.006.
2017
Haeberle S*, Raker V*, Haub J, Kim OK, Weng SY, Yilmaz OK, Enk AH, Steinbrink K, Schuppan D, Hadaschik EN. Regulatory T cell deficient scurfy mice exhibit a Th2/M2-like inflammatory response in the skin. J Dermatol Sci. 2017 Sep;87(3):309-311
Salz M, Haeberle S, Hoffmann J, Enk AH, Hadaschik EN. Elevated IL-31 serum levels in bullous pemphigoid patients correlate with eosinophil numbers and are associated with BP180-IgE., J Dermatol Sci. 2017 Sep;87(3):285-291
2015
Hadaschik EN, Wei X, Leiss H, Heckmann B, Niederreiter B, Steiner G, Ulrich W, Enk AH, Smolen JS, Stummvoll GH. Regulatory T cell-deficient scurfy mice develop systemic autoimmune features resembling lupus-like disease. Arthritis Research & Therapy, 2015 17:35
DOI 10.1186/s13075-015-0538-0
2010
Huter EN, Natarajan K, Torgerson T, Glass DD and Shevach EM. Autoantibodies in Scurfy mice and IPEX patients recognize Keratin 14. J. Invest. Dermatol. 2010; 130:1391-9.
2009
Huter EN, Stummvoll GH, DiPaolo RJ, Glass DD, Shevach EM. Cutting Edge: Antigen-specific TGFb-induced Regulatory T cells Suppress Th17-Mediated Autoimmune Disease. Int. Immunopharmacol. 2009; 9:540-45.
2008
Huter EN, Stummvoll GH, DiPaolo RJ, Glass DD, Shevach EM. Cutting Edge: Antigen-specific TGFb-induced Regulatory T cells Suppress Th17-Mediated Autoimmune Disease. J. Immunol. 2008; 181:8209-13.
Huter EN, Punkosdy GA, Glass DD, Cheng LI, Ward JM, Shevach EM. TGF-beta-induced Foxp3+ regulatory T cells rescue scurfy mice. Eur. J. Immunol. 2008; 38:1814-21.