



Congenital Disorders of Glycosylation (CDG)
Arbeitsgruppenleitung: Priv.-Doz. Dr. rer. nat. Christian Thiel
Die Glykosylierung von Proteinen und Lipiden (Anheftung von Zuckerstrukturen an ein Eiweiß- oder Fettmolekül) spielt bei einer Vielzahl von biologischen Prozessen eine wichtige Rolle. Angeborene Defekte, die die Synthese oder die Prozessierung von Zucker- (Glykan-) strukturen von Glykokonjugaten betreffen, führen beim Menschen zumeist zu schweren neurologischen Defekten, die als "Congenital Disorders of Glycosylation" (CDG = angeborene Defekte der Glykosylierung) bezeichnet werden. Diese auf ein Protein übertragenen Zuckerketten gewährleisten u.a. die Funktionalität der Proteine beispielsweise als Enzyme, greifen weiterhin auch in eine Vielzahl lebenswichtiger Prozesse wie Wachstum, Differenzierung, Entwicklung von Organen, Signalübertragung, Abwehr, Entzündung sowie maligne Entartung ein.
Bei CDG-Patienten ist jeweils einer der zahlreichen Schritte, die in der Zelle die Zuckeranheftung steuern, defekt. Nachfolgend kommt es bei den Patienten oftmals zu einer Vielzahl von Symptomen wie bspw. Gedeih- und Entwicklungsverzögerungen, Durchfälle, Leber-, Herz- und Nierenprobleme, Blutgerinnungsstörungen, Krampfanfälle, verringertes Kleinhirnvolumen, Muskelschwäche, Fettpolster an Oberarmen und Gesäß, invertierte Brustwarzen, allgemeine geistige Retardierung, verzögerte Sprachentwicklung, Müdigkeit und Schwäche.
Obwohl seit der molekularen Erstbeschreibung von Patienten mit einem Glykosylierungsdefekt im Jahr 1996 die genetischen Ursachen von einer Vielzahl weiterer CDG-Defekte aufgeklärt werden konnten (Stand 04/ 2022: 165 verschiedene monogenetische Defekte), steht zu vermuten, dass die bislang weltweit bekannten Patienten lediglich die Spitze eines Eisbergs darstellen.
CDG zählt zu den seltenen Stoffwechselerkrankungen.
Glykosylierung von Proteinen und Lipiden
Die Glykosylierung stellt eine der häufigsten Formen der Modifikation von Proteinen und Lipiden in Tieren, Pflanzen und Bakterien dar. Glykokonjugate treten in subzellulären Organellen wie dem endoplasmatischen Retikulum, dem Golgi, den Lysosomen, den Peroxisomen aber auch im Cytoplasma auf. Sie sind sowohl in zellulären Membranen als auch in extrazellulären Flüssigkeiten und Matrices vertreten.
Insbesondere Glykoproteine greifen in eine Vielzahl lebenswichtiger Vorgänge wie Wachstum, Differenzierung, Entwicklung von Organen, Signalübertragung, Abwehr, Entzündung und maligne Entartung ein. Im Rahmen der Glykoproteinbiosynthese werden Kohlenhydratketten schrittweise durch eine Reihe von Glykosyltransferasereaktionen erzeugt und anschließend kovalent an Aminosäurereste eines Glykoproteins angeheftet.
Bei der Glykosylierung von Proteinen wird zwischen der N-Glykosylierung und der O-Glykosylierung mit ihren vielen verschiedenen Untergruppen (u.a. O-GalNAcylierung, O-GlcNAcylierung, O-Fukosylierung oder den alpha-Dystroglykanopathien) unterschieden. Daneben gibt es noch die Glycosphingolipid-, die Glycosylphosphatidylinositol (GPI)-Anker- sowie Glycosaminoglykosylierung.
Mehr als die Hälfte der bislang bekannten CDG-Defekte entfallen auf den Bereich der N-Glykosylierung, wobei PMM2-CDG mit 60-70% aller Erkrankten zudem den mit Abstand häufigsten Defekt aller Glykosylierungsarten repräsentiert.
Ablauf der N-Glykosylierung
Die Synthese von Glykoproteinen ist ein höchst komplexer Vorgang, der im Cytoplasma, an der cytosolischen Seite der Membran des Endoplasmatischen Reticulums (ER), im Lumen des ER und im Golgi-Apparat abläuft. Die N-Glykosylierung beginnt an der cytosolischen Seite der ER-Membran, wo schrittweise zwei N-Acetylglucosamin (GlcNAc)- und fünf Mannose-Reste (Man) auf das Lipid Dolicholphosphat übertragen werden, so dass das Intermediat Man5GlcNAc2-PP-Dolichol entsteht. Dieses Intermediat wird nun in das Lumen des ER geflippt. Hier erfolgt die Übertragung von vier weiteren Mannoseresten sowie drei Glucoseresten (Glc), so dass das vollständige Oligosaccharid `G3Man9GlcNAc2-PP-Dolichol´ entsteht. Die Oligosaccharyltransferase (OST) transferriert das Oligosaccharid anschließend „en bloc“ auf die Asparaginreste von wachsenden Polypeptiden. Noch während sich das Protein in seine richtige Konformation faltet, werden die Glucosereste sowie ein Mannoserest von zwei Glucosidasen und einer Mannosidase wieder abgespalten. Das Protein wird in den Golgi-Apparat transportiert, wo weitere Trimming und Elongationsschritte mit weiteren Zuckerresten (N-Acetylglucosamin, Galaktose, Fucose, Sialinsäure) stattfinden.
Die einzelnen CDG-Typen werden anhand des jeweils betroffenen Proteins zusammen mit dem Kürzel `-CDG´ voneinander unterschieden. Ein Defekt in der Phosphomannomutase 2 (PMM2) wird daher als `PMM2-CDG´ bezeichnet.
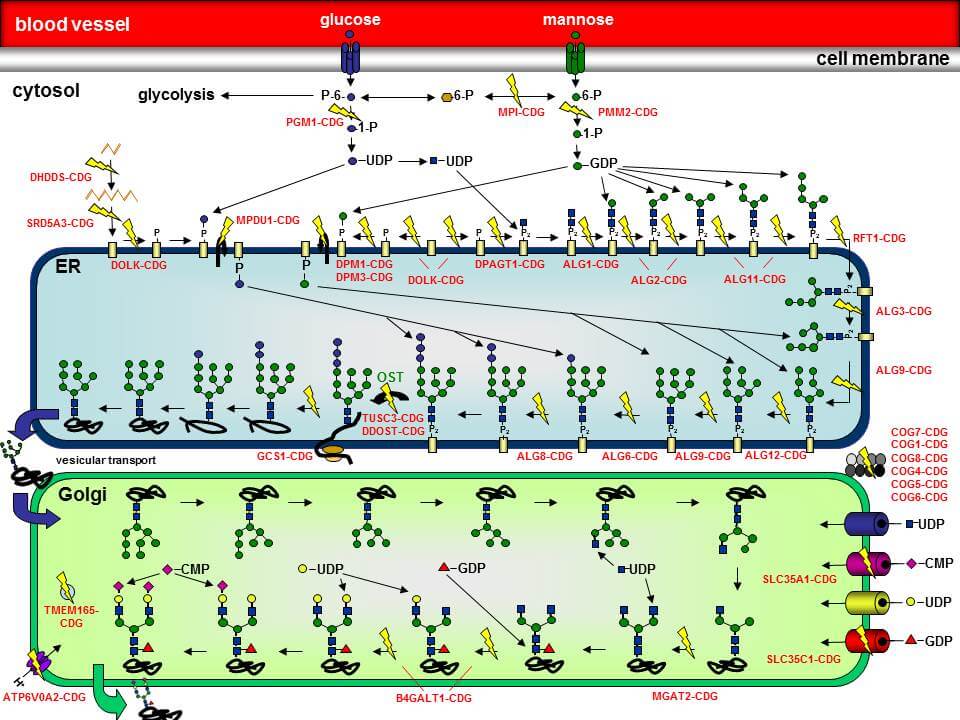
In der Abbildung sind eine Vielzahl der bisher identifizierten N-Glykosylierungsdefekte durch gelbe Blitze hervorgehoben. Der Name des jeweiligen Defekts ist ebenfalls angegeben.
Symbole
Einleitende Untersuchung bei CDG-Verdacht
CDG-Verdachtspatienten werden im Allgemeinen zunächst auf eine Veränderung der N-Glykosylierung hin untersucht. Weltweit am häufigsten verbreitet ist hier die Durchführung einer isoelektrischen Fokussierung (IEF) anhand des Serummarkers Transferrin. Dieses Glykoprotein trägt zwei biantennäre sialylierte Oligosaccharide vom komplexen Typ, die aufgrund der terminalen Sialinsäurereste negativ geladen sind. Fehlen aufgrund eines Glykosylierungsdefekts des Patienten eine oder beide Ketten, kommt es zu einer Ladungsverschiebung und folglich zu einer Veränderung des isoelektrischen Punktes des Proteins. Diese Verschiebung kann dann mit Hilfe der IEF nachgewiesen werden (siehe Abbildung IEF).
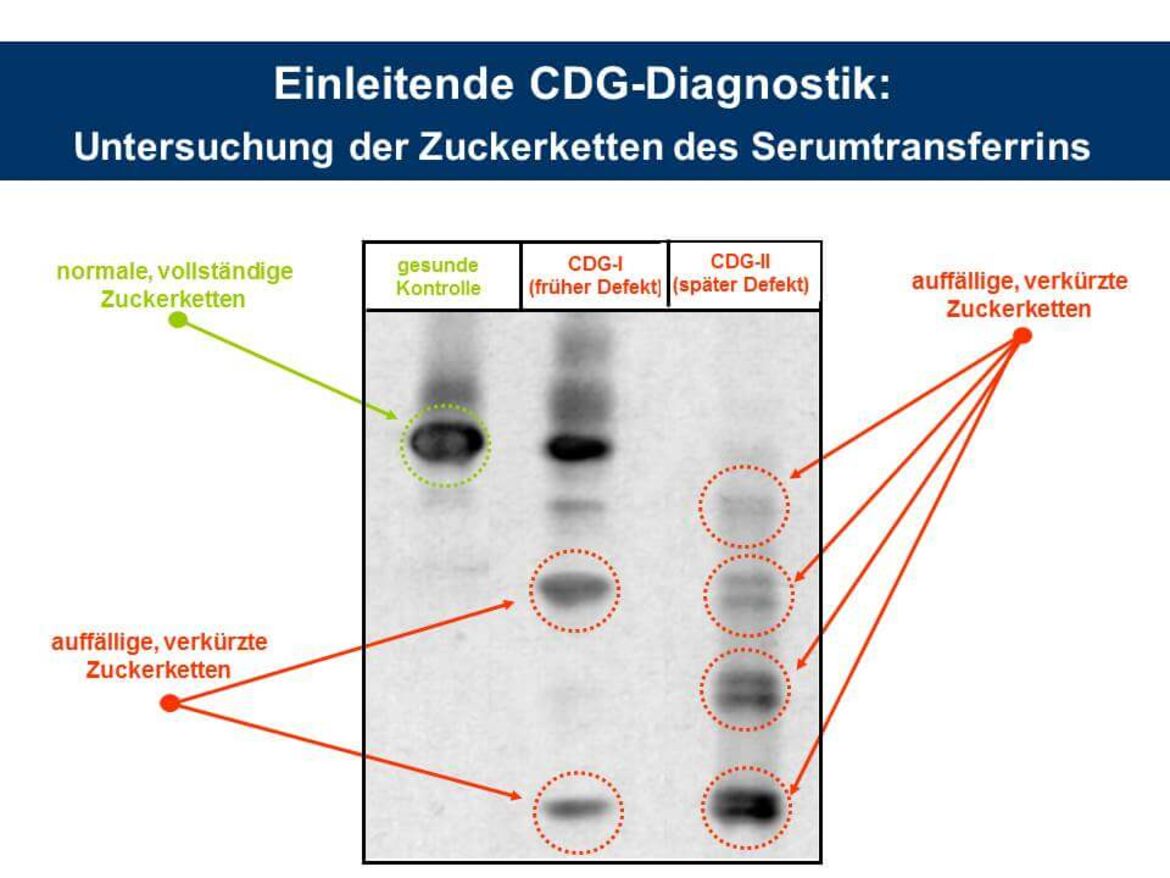
Hinweis: Das vollständige Glykosylierungsmuster des Serum-Transferrins wird zum Teil erst in einem Alter von 3 Monaten ausgeprägt. Daher wird bei Patienten, die jünger als 3 Monate sind, ein alternatives Markerprotein der CDG-Diagnostik, das alpha-1-Antitrypsin, auf eine Glykosylierungsveränderung hin analysiert.
Allerdings lassen sich CDG-Defekte hier nur in frühe (CDG-I) und späte Erkrankungstypen (CDG-II) unterteilen. Zudem lassen sich auch nicht alle N-Glykosylierungsdefekte anhand des Serumtransferrins erkennen. Wesentlich genauere Ergebnisse können wir in unserem Labor mit der Durchführung einer massenspektrometrischen Analyse der N-Glykane aller Glykoproteine aus dem Serum erzielen. Hierdurch ist es uns zum Teil sogar möglich, einen CDG-Defekt anhand seines spezifischen Glykanprofils in der Serumprobe zu identifizieren (z.B. PMM2-CDG, Abbildung MS).
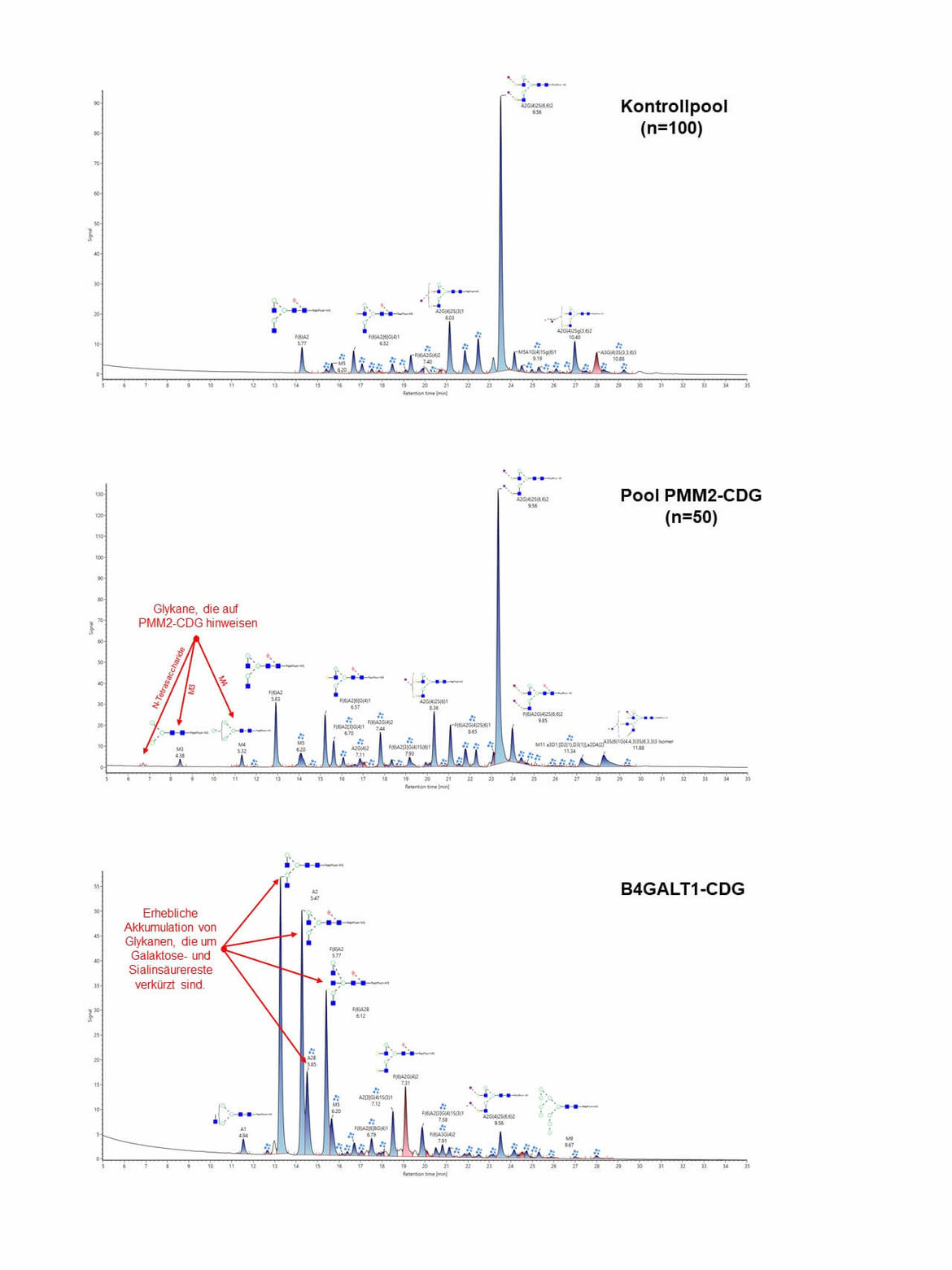
Massenspektrometrische Untersuchung der N-Glykane von Gesamtserumglykoproteinen
Nach Abspaltung der Zuckerketten (N-Glykane) von Gesamtglykoproteinen aus dem Serum mittels PNGase F wurden diese massenspektrometrisch aufgetrennt und analysiert. Bei PMM2-CDG zeigen sich spezifische Akkumulationen von N-Glykanen (N-Tetrasaccharid, M3 und M4) wie sie bei PMM2-CDG zu finden sind.
Bei einem Defekt in der Golgi Beta-1,4-Galaktosyltransferase (B4GALT1-CDG) ergeben sich Ansammlungen von Glykanen, die um Galaktose- und nachfolgend auch um Sialinsäurereste verkürzt sind.
Um diese ersten Untersuchungen durchführen zu können, benötigen wir lediglich eine kleine Menge an Serum (ca. 0,5ml). Einsendung von Plasma oder Trockenblutkarten bitte nur nach vorheriger Absprache.
Weitere häufig genutzte Markerprotein in der CDG-Diagnostik sind das Apoliprotein CIII zur Erkennung von O-Glykosylierungsdefekten vom Core-1-Mucintyp oder die alkalische Phosphatase, die bei GPI-Ankerdefekten oftmals auffällig ist. Nicht für alle Glykosylierungsarten gibt es allerdings etablierte Markerproteine im Blut, so dass hier vor allem auf Zellkulturstudien ausgewichen werden muss.
Wichtig: Im Vorfeld sollten vom Einsender Alkoholabusus, klassische Galaktosämie, Fruktoseintoleranz sowie bakterielle oder virale Infektionen ausgeschlossen werden.
Weiterführende Laboruntersuchungen
Wird während der CDG-Diagnostik eine Auffälligkeit erkannt, finden weiterführende Untersuchungen statt, um den Glykosylierungsdefekt näher einzugrenzen oder sogar zu identifizieren. Diese können genetisch über eine Paneldiagnostik, Whole Exome oder Next Generation Sequenzierung erfolgen. Biochemische Analysen umfassen beispielsweise die Aktivitätsbestimmung von Enzymen und Zuckertransportern oder Studien in der Zellkultur.
Ob genetisch oder biochemisch mit der weiterführenden Diagnostik begonnen wird, ist unerheblich. Wichtig ist, dass sich genetische und biochemische Untersuchungen ergänzen sollte, um falsch positive Befunde für die Patienten und deren Familien zu vermeiden.
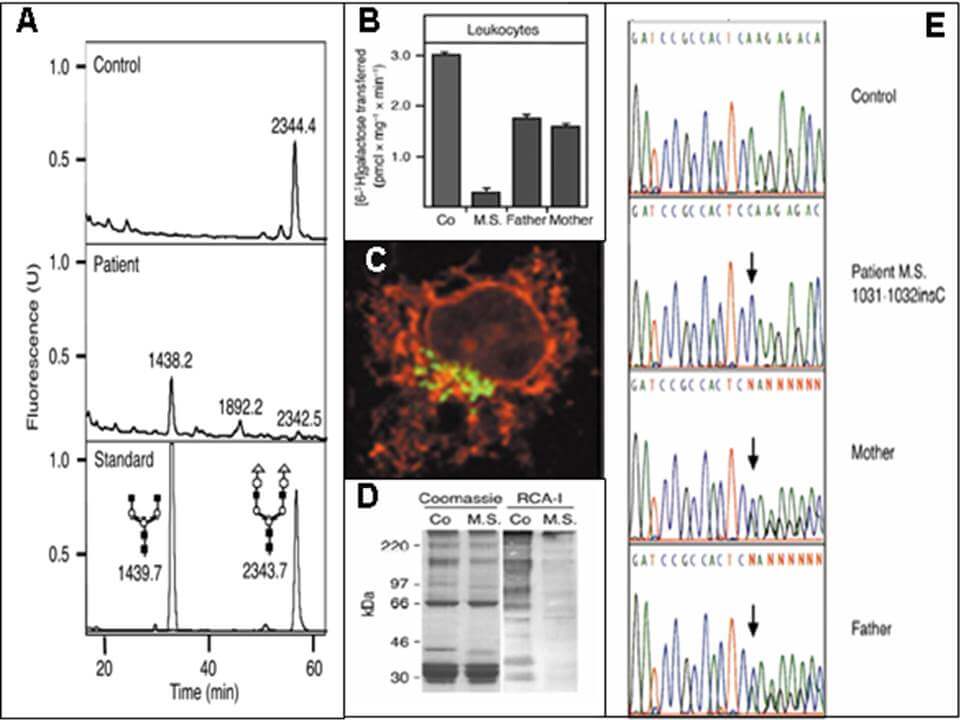
Weiterführende Laboruntersuchungen beinhalten bspw. die Analyse von N-Glykanen aus Serum, Plasma oder Trockenblutkarten mittels Massenspektrometrie (A), Messungen von Enzym- bzw. Transporteraktivitäten (B), Immuncytochemie (C), Lektinbindestudien (D) sowie molekularbiologische Analysen (E) in Patientenfibroblasten bzw. -lymphoblasten.
PMM2-CDG
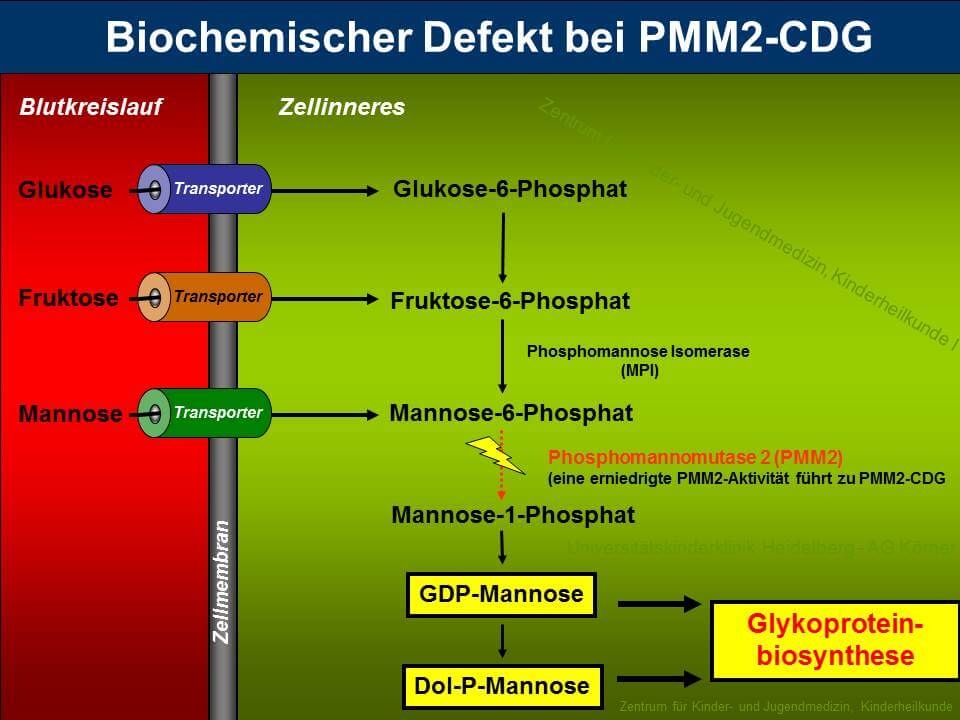
PMM2-CDG (zuvor CDG-Ia) ist sowohl der am längsten bekannte als auch der am häufigsten vorkommende Typ unter den "Congenital Disorders of Glycosylation". Etwa 60% aller CDG-Patienten leiden unter dieser Erkrankung, die durch die verminderte Aktivität des Enzyms Phosphomannomutase 2 (PMM2) hervorgerufen wird. PMM2 setzt im Cytosol Mannose-6-Phosphat zu Mannose-1-Phosphat um, welches für die Synthese von GDP-Mannose, GDP-Fucose und Dolichol-Phosphat-Mannose benötigt wird (siehe Abbildung 3). Durch die verminderte Bereitstellung von GDP-Mannose und Dolichol-Phosphat-Mannose kommt es bei den Patienten zu einer Verkürzung der Dolichol-verknüpften Oligosaccharide, die ein schlechtes Substrat für die Oligosaccharyltransferase darstellen, was zum partiellen Verlust kompletter Zuckerketten auf reifen Glykoproteinen führt.
Die verminderte Enzymaktivität der PMM2 konnte auf Mutationen im PMM2-Gen zurückgeführt werden, das beim Menschen auf Chromosom 16p13 lokalisiert ist. Auffällig ist, dass bei allen bisher identifizierten Mutationen eine Restaktivität von PMM2 beobachtet wurde, was Anlass zu der Vermutung gibt, dass ein totaler Aktivitätsverlust der Phosphomannomutase unvereinbar mit dem Leben ist.
Therapien und therapeutische Ansätze
Obwohl weltweit von klinischen und biomedizinischen Arbeitsgruppen stark geforscht wird, bestehen für die meisten CDG-Typen nur symptomatische Behandlungsmaßnahmen.
Einfache und gut verträgliche Therapiemöglichkeiten liegen aber bereits für folgende CDG-Defekte vor:
MPI-CDG (früher CDG-Ib; orale Mannosegabe. Hier aber im Vorfeld unbedingt die Restaktivität der betroffenen Phosphomannose Isomerase bestimmen lassen)
PGM1-CDG (Galaktosegabe)
CAD-CDG (orale Uridingabe)
GFUS-CDG (orale Fukosegabe)
GNE-CDG (Orale ManNAc-Gabe)
SLC35C1-CDG (orale Fukosegabe)
FUT8-CDG (orale Fukosegabe)
SLC35A2-CDG (orale Galaktosegabe)
SLC39A8-CDG (orale Galaktose- und Mangangabe)
TMEM165-CDG (orale Galaktosegabe)
Weitere Therapien, auch zum häufigsten CDG-Defekt (PMM2-CDG), sind in der Erprobungsphase.
Aktuell suchen wir CDG-Patienten (Defekt innerhalb der N-Glykosylierung und/ oder O-Mucintypglykosylierung), die Interesse an der Einnahme eines bestimmten Vitamins haben, das bei einem vorliegenden Glykosylierungsdefekt oftmals nicht in ausreichender Menge bereitgestellt werden kann.
Bei Interesse nehmen Sie bitte Kontakt mit uns auf: Christian.Thiel(at)med.uni-heidelberg.de.
Forschung
Neben der Identifizierung neuer, bisher unbekannter Defekte des CDG-Typs sind wir besonders an der Aufklärung pathophysiologischer Vorgänge bei CDG und den damit verbundenen Einblicken in die generelle Bedeutung der Proteinglykosylierung sowie an der Entwicklung therapeutischer Strategien für CDG interessiert. So haben wir in den letzten Jahren die molekularen Ursachen für eine ganze Reihe von CDG-Typen identifiziert und verschiedene Tier- und Zellkulturmodelle generiert und charakterisiert. Durch die apparative Ausstattung unserer Labore können sowohl die eingehenden Patienten- als auch Forschungsproben umfassend biochemisch, genetisch, zellbiologisch oder histochemisch im eigenen Haus analysiert werden.
Unsere Forschungsprojekte werden durch Sachbeihilfen öffentlicher Drittmittelgeber aus Deutschland sowie der Europäischen Union gefördert.
Mitarbeiter
Arbeitsgruppenleiter
-

PD Dr. rer. nat. Christian Thiel
Schwerpunkt
Congenital Disorders of Glycosylation (CDG)/ Funktionsbereich Glykosylierungsstörungen (CDG)
Biologiedoktorand
Med.-techn. Laborassistenten/-innen
-

Virginia Geiger
-

Karolin Schaefer
Bio.-Techn. Assistentin
-
Simone Hengst
Postdoc
Praktikantin
-
Fabienne Rech
